Objective: To investigate time to maximal platelet inhibition after an oral loading dose of ASA. The effect of ex vivo reversal platelet inhibition by desmopressin (DDAVP) was also studied.
Methods: Ten healthy volunteers were given a 300 mg ASA-tablet. Blood was sampled at 0, 15, 30, 60, 120 and 180 minutes. DDAVP was added to the samples taken at 120 minutes.
Samples were analysed with a Multiplate® platelet aggregometer (MEA) using arachidonic acid (AA), collagen and thrombin aggregation agonists.
Results: Platelet inhibition was observed in the sample activated by AA at 15 minutes but not until 120 minutes in the samples activated by collagen. No platelet inhibition was seen in the samples activated by thrombin. The median time to maximal AA-induced platelet inhibition of <30 U was 30 (interquartile range 15-90) minutes. Ex vivo DDAVP did not reverse platelet inhibition. Subgroup analysis did not show any gender differences.
Conclusions: ASA induces a strong platelet inhibition within 30 minutes of oral ingestion, with no gender differences. Ex vivo DDAVP did not reverse ASA’s platelet inhibition.
Aspirin, desmopressin, platelet aggregation, healthy volunteers, gastric absorption
Acetylsalicylic acid (ASA) is frequently prescribed in the treatment and prevention of acute coronary syndrome (ACS) and acute ischaemic stroke, even if its role in primary prevention is unclear [1,2]. ASA is also used as an over-the-counter drug for pain and inflammation. ASA- associated haemorrhage risk is an issue regardless of indication [3,4] since it at low doses (75-150mg daily) inhibits platelets, primarily by irreversible inhibition of platelet cyclooxygenase (COX), preventing the formation of thromboxane A2 (TXA2) and thereby, diminishing platelet aggregation. At higher ASA doses, also prostacyclin is inhibited, counteracting the ASA low dose antithrombotic effect [5]. In emergency situations ASA therapy is often started with a loading dose of 300 mg [6].
Standard aspirin therapy is currently often combined with a P2Y12-antagonist, so called dual antiplatelet therapy in coronary care (DAPT), with strengthened platelet inhibition [7]. There is a variable individual response both to ASA and P2Y12-antagonists, [8] highlighted by the introduction of point-of-care (POC) platelet function devices that are easy to use and rapidly show results [9]. Examples of POC platelet function devices are PFA-100, VerifyNow, PlateletWorks, TEG-PM (Platelet Mapping), ROTEM platelet and Multiplate Electrode Aggregometry (MEA). Several studies have measured ASA responsiveness with these devices.
MEA has been used for studies of platelet aggregation in different clinical settings [10,11]. MEA reliably measures ASA platelet inhibition and is comparable to standard light transmission aggregometry and other POC platelet function devices [12]. In a study by Karon et al. MEA was the best test for measuring the effects of aspirin at the initiation of treatment as compared to TEG-PM and VerifyNow [13]. It is important to measure the baseline platelet function with MEA prior to start of ASA as MEA can show platelet inhibition despite no ASA intake [14]. No study has yet investigated the time to peak platelet inhibition as measured by MEA in healthy volunteers after a normal per-oral loading dose of ASA (300-325 mg): previous studies have investigated either very early inhibition in patients with acute myocardial infarction [15] or a lower ingested dose [16].
Desmopressin (DDAVP) raises the plasma levels of factor VIII and von Willebrand factor (vWF) by stimulating their release from von Weibel Palade bodies on endothelial cells [17]. DDAVP is used clinically in the treatment of mild haemophilia A and von Willebrand disease, in coagulopathies associated with hepatic cirrhosis and uraemia, and various congenital and acquired causes of platelet dysfunction [18]. Desmopressin has also been used to reverse ASA effects on platelets [19]. DDAVP has also been proposed to have direct effects on platelets, but in a study from our laboratory, MEA failed to demonstrate any in vitro effect of desmopressin on whole blood platelet aggregation in healthy volunteers with no platelet inhibiting drugs [20].
The primary aim of this study was to investigate the time to peak platelet inhibition measured with MEA, in blood from non-fasting healthy volunteers after an oral loading dose of 300 mg of ASA. A secondary aim was to investigate if the ex-vivo addition of DDAVP reversed the ASA-induced platelet inhibition.
This prospective observational study was approved by the Swedish Ethical Review Authority (registration numbers DNR 2010/482) and was conducted in accordance with the World Medical Association Code of Ethics (Helsinki Declaration 1975). The study was performed in 2019 at the Department of Intensive and Perioperative Care, Skåne University Hospital, Lund, Sweden. The manuscript was prepared in accordance with the STROBE guidelines for observational studies.
Ten healthy non-smoking volunteers without any medication 14 days prior to inclusion, were recruited and included after informed and signed consent. Five females and five males were studied, balancing gender effects on platelet function [21].
To avoid diurnal variation in platelet function, the volunteers were instructed to eat a normal breakfast including 1 bowl of yoghurt with cereals and one or two pieces of bread, before starting the study at 8 am [22]. During the study only ingestion of tea, coffee or water was allowed. In a recent study conducted by Krekels et al. no significant effect on platelet aggregation was seen after coffee, high-fat meals, or physical exercise [23].
The volunteers were given a single 300mg oral loading dose of non-enteric ASA (4 tablets of Trombyl® 75mg, Pfizer, New York, USA). The tablets were swallowed with water, without being chewed, since chewing ASA tablets has been shown to increase absorption and shorten the time to a 50% reduction of serum thromboxane B2 (TXB2) concentration from 12 to 5 minutes [24]. Enteric coated ASA tablets have poorer absorption from the higher pH environment of the small intestine and may result in less platelet inhibition [25].
A peripheral vein catheter (PVC) (Venflon™, BD, Plymouth, UK) was inserted in an antecubital vein. Venous blood was drawn from the PVC using the Vacutainer System® (BD, Plymouth, UK) immediately before and then at 15, 30, 60, 120 and 180 minutes after ingestion of ASA. The first tube for each sampling was discarded before blood for MEA analysis was drawn using a 3 ml hirudin tube (Roche Diagnostics Scandinavia AB, Bromma, Sweden). The samples were incubated for 30 minutes at room temperature before analysis as recommended by the manufacturer [26] and Würtz et al. [27].
MEA analysis was performed using the Multiplate® analyser (Roche Diagnostics Scandinavia AB, Bromma, Sweden). The analysis is based on whole blood and measures the change in electric impedance between two electrodes over time upon addition of a platelet activation agonist. Activated platelets stick to the electrodes causing the impedance to increase with increased platelet aggregation. This change in electrical current is plotted in a graph as aggregation units (AU) against time (min). The area under the curve (AUC) represents the extent of platelet aggregation and is expressed as units (U) where 1 U = 10 AU*min. Three tests with different platelet agonists were performed on each sample. The agonists used were ASPItest® with a test concentration of 0.5 mM of arachidonic acid, which initiates platelet aggregation through the COX pathway; TRAPtest® with 32 µM thrombin receptor activating peptide; and COLtest® with 3,2 µg/ml of collagen as described in the manufacturer’s instructions [26]. Maximal ASA-effect as measured with the ASPItest was defined as an AUC of <30 U in accordance with the manufacturer’s documentation based on studies using hirudin anticoagulated test tubes [28,29].
In addition to the analyses described above, an extra sample was taken 2 hours after ASA ingestion to investigate whether DDAVP added in vitro attenuates the effect of ASA as measured with MEA. The samples were incubated at room temperature for 30 minutes. Each sample was divided in two aliquots with 1.3 ml of blood in each. One of the aliquots was mixed with 0.1 ml of DDAVP with a concentration of 12.6 ng/ml (Octostim®, Ferring Copenhagen, Denmark) to a target concentration of 0.9 ng/ml. This concentration is higher than the average maximal plasma concentration in vivo after a normal dose (0.3 µg/kg) of DDAVP, which is 0.6 ng/ml [30]. The other aliquot was mixed with 0.1 ml of normal saline (Fresenius Kabi, Bad Homburg, Germany) to serve as a control. Both aliquots were then analyzed with MEA using ASPItest®, TRAPtest® and COLtest® as described above.
Statistical analysis was done using GraphPad Prism version 8.4.2 for Windows (GraphPad Software, La Jolla, California, USA). All variables were considered non- parametric and were presented as median with (interquartile range). Friedman’s test was used to compare MEA AUC values at baseline (0 minutes) with the different blood sampling times (15, 30, 60, 120 and 180 minutes). Wilcoxon’s two-tailed matched-pairs rank sum test was used to compare samples taken at each blood sampling time to the baseline, as well as comparing DDAVP ex-vivo spiked blood samples to non-DDAVP spiked samples. A p-value of <0.05 was considered significant. As gender has been described to affect platelet function, [20] a subgroup analysis with a Mann Whitney test of gender differences was also performed, analysing Delta differences between before ASA and at 120 minutes for ASPItest®, COLtest® and ASPI® with DDAVP.
The median age of the 10 volunteers was 31 years with a range of 21-53 years.
Time to maximal effect after oral ingestion of ASA: MEA AUC using ASPItest® decreased significantly after the loading dose of 300 mg ASA (p<0.001) (Figure 1). The median MEA AUC using ASPItest® before ASA ingestion was 99 U (Q1-Q3:94-103). At 15 minutes there was a significant decrease to 87 U (19-94 U), (p=0.006), and at 30 minutes there was a further decrease to 23 U (19-81 U), (p=0.002). The median of all subsequent measures was below the cut-off value for maximal platelet inhibition of 30 U. The median time to maximal ASA-induced platelet inhibition measured with ASPItest® test was 30 minutes (15-90 minutes). All but 2 of the 10 volunteers showed maximal platelet inhibition on ASPItest® within 60 minutes after ASA ingestion. One was a 53-year-old male reaching <30 U at 180 minutes; the other a 31-year-old male reaching <30 U at 120 minutes. Three of the volunteers already showed maximal platelet inhibition at the first sampling at 15 minutes: 2 females both 21 years old and one male 23 years old. On COLtest® the decrease in platelet aggregation compared to baseline was not significant at 15, 30 or 60 minutes. There was, however, a statistically significant decrease from baseline to 120 minutes (p=0.002) and 180 minutes (p=0.002) (Figure 2).
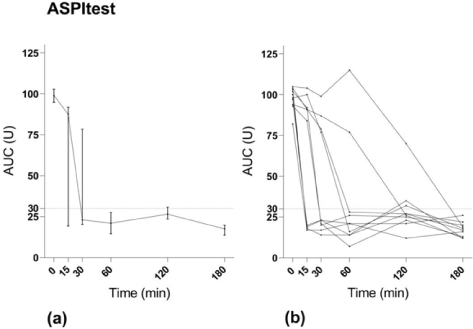
Figure 1. MEA results with ASPItest. (a) Median values with interquartile range. (b) Values for individual study subjects.
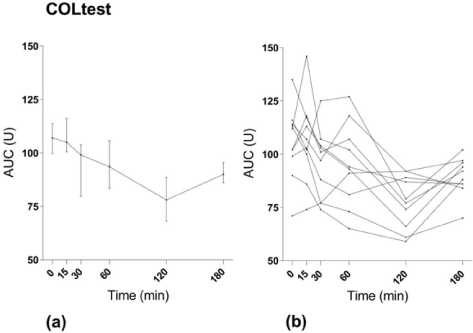
Figure 2. MEA results with COLtest. (a) Median values with interquartile range. (b) Values for individual study subjects.
TRAPtest showed no significant difference between MEA AUC at baseline and sampling after ingestion of ASA (p=0.27) (Figure 3).
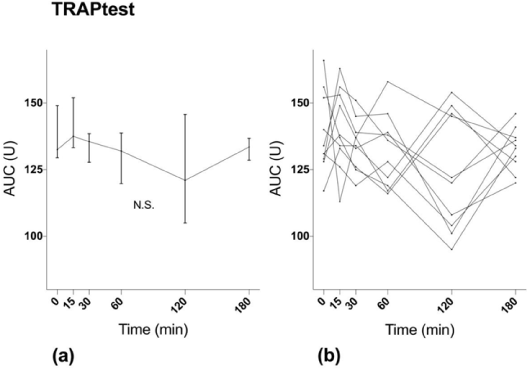
Figure 3. MEA results with TRAPtest. (a) Median values with interquartile range. (b) Values for individual study subjects.
The addition of DDAVP ex vivo did not increase the MEA AUC using ASPItest® compared to the controls (Figure 4) or the other MEA tests (Table 1).
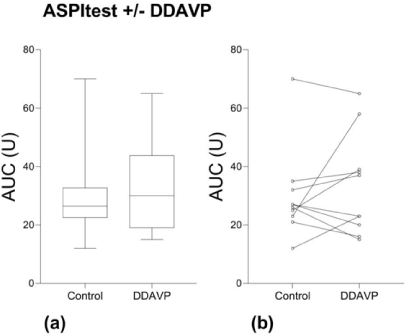
Figure 4. MEA with arachidonic acid activation (ASPItest) with and without DDAVP. (a) Median, Q1-Q3, min-max. (b) Values for individual study subjects.
Table 1. Effects of ex vivo added DDAVP. All values are presented as median (interquartile range).
A subgroup analysis of gender differences did not detect any significant differences in Delta response to ASA (before and at 120 minutes after the loading dose) for ASPItest®: p= 0.14; COL: p= 0.06 and ASPItest®-DDAVP: p=0.11.
In this prospective observational study, a peroral loading dose of 300 mg (non-chewed) ASA to healthy volunteers induced demonstrable and significant platelet inhibition within 30 minutes with considerable interindividual differences. Ex vivo desmopressin did not reverse this platelet inhibition.
Zeymer et al. studied platelet inhibition with the MEA ASPItest® 5 and 20 minutes after patients with suspected or confirmed acute myocardial infarction were given either 250 mg or 500 mg of intravenous (iv) ASA or 300 mg oral chewed ASA tablets [15]. They also measured arachidonic acid (AA) induced thromboxane B2 (TxB2) synthesis as a gold standard marker of COX-1 inhibition by ASA. The baseline mean ASPItest® AUC was 86 U. Maximal MEA platelet inhibition was on average already reached at 5 minutes when ASA was given intravenously (mean±SD ASPItest® AUC 20.6±12 for 250 mg and 23.0±19 for 500 mg respectively) with no significant changes thereafter. In the per-oral group the platelet inhibition was on average minimal (mean±SD ASPItest® AUC 75.6±39) at 5 minutes and still sub-maximal (mean±SD ASPItest® AUC 42.5±34) in the peroral group at 20 minutes.
Schwertner et al. investigated early pharmacokinetics and platelet inhibition by three different oral formulations of ASA in healthy fasting volunteers using optical aggregometry on platelet-rich plasma. Platelet inhibition was defined as a 50% decrease in optical density, which occurred in all volunteers within 25 minutes [31]. Feldman et al. studied early pharmacokinetics of ASA and changes in serum TxB2 concentration after oral administration of loading doses of 325 mg in three different formulations, to twelve healthy, fasting volunteers [24]. They found a 90% decrease in TxB2 from the baseline after a 25.7±4.6 min for buffered ASA tablets swallowed intact, 13.6±1.9 min for chewed buffered tablets and 15.8±1.8 min for buffered tablets dissolved in water (all mean±SD).
The results of our study seem to corroborate the above studies regarding the approximate time to maximal platelet inhibition after oral ASA administration. There were considerable inter- individual variations among our volunteers, with six of the participants responding with maximal platelet inhibition within half an hour, whereas two of the participants had not reached full inhibition until after 2 and 3 hours, respectively. Due to our small sample size, it is not possible to say if this accurately reflects the proportion of the population who have a delayed response to oral ASA, or whether variations depend on age or gender. Rubak et al. could not demonstrate a relationship between age and MEA ASPItest® and COLtest® AUC in 121 healthy volunteers between 17 and 66 years [32]. MEA AUC was higher in females, probably reflecting estrogen effects on platelets. In our subgroup analyses we could not show any gender differences in baseline values or in the ASA or ex vivo DDAVP responses, but our study is underpowered for this.
The results from the study of Zeymer et al. indicate that gastrointestinal absorption is an important factor affecting the onset of action of ASA-mediated platelet inhibition [15]. Of note is that the baseline median ASPItest® platelet aggregation was higher in our study (99 U (94- 103) than in the patients studied by Zeymer et al. (mean AUC 86 U)). This may be a result of different baseline characteristics of the study subjects. Zeymer et al. excluded patients who had taken ASA or NSAIDs within the previous 48 hours, as opposed to two weeks in our study. Peerschke et al. studied blood collected in hirudin anticoagulated tubes from 193 healthy volunteers, and found a mean±SD baseline ASPItest® AUC of 103.4 ±24 (median 105), ie similar to our results [33].
Several studies have identified a variable patient response to ASA [34,35] which some researchers have referred to as an ASA resistance. This is an ill-defined term, as are the mechanisms behind it [36]. Comparison of different point of care (POC) and laboratory tests demonstrates platelets to be inhibited to different degrees by different drugs [34,37]. Procoagulant activity and critical illness can also affect platelet function and decrease the response to ASA [35,38]. The variable ASA response could also depend on COX genetic variants [39]. Recently the terminology “high on-aspirin (residual) platelet activity” has been introduced [40].
The ASA effect on MEA active by collagen (COLtest®) has been described earlier [12,41]. TRAPtest® is a very strong activator containing thrombin receptor activating protein, and usually there is no inhibitory response to even strong platelet-inhibiting drugs [15].
Our study adds to the knowledge of when to expect maximal platelet inhibition after an oral loading dose of ASA measured with MEA. It is not yet known if the difference between onset of action of intravenous versus oral ASA has any clinical significance with regards to morbidity and mortality in acute ischemic stroke or acute myocardial infarction.
Earlier studies suggest a clinically significant decrease in mortality from acute myocardial infarction with earlier (within a median of 1.6 hours) versus later (median 3.5 hours) administration of oral ASA after onset of symptoms [42]. Our results indicate that in healthy non-fasting individuals there seems to be a considerable variation in the onset of action of platelet inhibition, perhaps reflecting a difference in gastric emptying and enteral uptake that is more pronounced than in fasting subjects. It would be of interesting to explore intra-individual differences in a cross-over study investigation of the onset of platelet inhibition between intravenous and per-oral administration in the fasting and non-fasting state. It might also be of interest to study platelet inhibition within the first hours after ASA administration to patients with suspected or confirmed acute coronary syndrome, at later intervals than previous studies with the aim of identifying any subgroup of patients with a considerably delayed onset of action of per-oral platelet inhibition: the clinical significance of finding such a subgroup would be being able to give intravenous rather than oral ASA.
There are some methodological issues concerning the MEA ASPItest®: whole blood MEA testing of samples collected in hirudin tubes has a higher variability than citrate, which is increased by ASA [33]. With ongoing ASA treatment, the MEA ASPItest® AUC has a coefficient of variation (cv) of 10 %, [12] but Pedersen et al. found a cv of 8% increasing to 48% in the ASPItest® before and after ASA treatment in healthy volunteers [43].
DDAVP may have a place in enhancing haemostasis in the presence of ASA-induced platelet inhibition [17-19,44]. This could be of interest in neurosurgery [45]. Otherwise, ASA should not be routinely withdrawn prior to surgery as it is considered a weak platelet inhibitor and this practice may cause thrombotic complications. The decision to withdraw ASA should be made considering risks and benefits for the individual patient and type of procedure [46].
In our study a clinically relevant concentration of DDAVP was ex vivo added to blood drawn at two hours after ASA ingestion. If this had had any reversal effect on ASA-mediated platelet inhibition it could not be measured with MEA. This is in line with a previous study by Pearson et al. that failed to demonstrate an effect of DDAVP added in-vitro [20]. Weber et al. used MEA with ASPItest® to investigate the effect of intravenously administered DDAVP in patients currently not treated with platelet inhibitors but who had suspected post-operative platelet dysfunction after coronary artery bypass grafting [47]. They found that DDAVP significantly enhanced platelet aggregation. Lethagen et al. found an ASA reversing effect for 3 hours after iv DDAVP with a glass bead platelet retention test [19]. The effect of DDAVP shows great interindividual variability in vWF/FVIII release [48]. VWF interacts with the glycoprotein IIb/IIIa (GPIIb/IIIa) platelet receptor, which is flow dependent for its activation, especially with high shear stress and rates as in the arterial circulation. No POC platelet test can detect this: special flow chamber techniques are needed [49].
In conclusion, a peroral loading dose of 300 mg (non-chewed) ASA induced demonstrable and significant platelet inhibition as measured by the Multiplate® platelet aggregometer, within 30 minutes. Ex vivo desmopressin did not reverse this platelet inhibition. There were considerable interindividual differences in the onset of peak platelet inhibition, which should be considered when studying ASA responsiveness. Desmopressin’s suspected reversal effect on decreased platelet function caused by ASA should be studied in vivo in future studies with advanced platelet function tests.
Department funding only.
The Authors declare(s) that there is no conflict of interest.
- Ganjehei L, Becker RC (2015) Aspirin dosing in cardiovascular disease prevention and management: an update. J Thromb Thrombolysis 40: 499-511.
- Stuntz M, Bernstein B (2017) Recent trends in the prevalence of low-dose aspirin use for primary and secondary prevention of cardiovascular disease in the United States, 2012–2015. Prev Med Rep 5: 183-186. [Crossref]
- Boyina KK, Nakkana G, Murthy VVR (2017) Buying Over-the-Counter Antacid Products Containing Aspirin? FDA Drug Safety Communication for Serious Bleeding Risk. Am J Gastroenterol 112: 654-655. [Crossref]
- Whitlock EP, Williams S, Burda BU, Feightner A, Beil T (2015) Aspirin use in adults: cancer, all-cause mortality, and harms: a systematic evidence review for the US Preventive Services Task Force. [Crossref]
- Louie S, Gurewich V (1983) The antithrombotic effect of aspirin and dipyridamole in relation to prostaglandin synthesis. Thromb Res 30: 323-335. [Crossref]
- Buerke M, Pittroff W, Meyer J, Darius H (1995) Aspirin therapy: optimized platelet inhibition with different loading and maintenance doses. Am Heart J 130: 465-472. [Crossref]
- Warlo EM, Arnesen H, Seljeflot I (2019) A brief review on resistance to P2Y 12 receptor antagonism in coronary artery disease. Thromb J 17: 11. [Crossref]
- Campbell J, Ridgway H, Carville D (2008) Plateletworks®. Molecular diagnosis & therapy 12: 253-258.
- Paniccia R, Priora R, Liotta AA, Abbate R (2015) Platelet function tests: a comparative review. Vasc Health Risk Manag 11: 133-148. [Crossref]
- Kander T, Dankiewicz J, Friberg H, Schött U (2014) Platelet aggregation and clot formation in comatose survivors of cardiac arrest treated with induced hypothermia and dual platelet inhibition with aspirin and ticagrelor; a prospective observational study. Crit Care 18: 495. [Crossref]
- Kander T, Larsson A, Taune V, Schött U, Tynngård N (2016) Assessment of haemostasis in disseminated intravascular coagulation by use of point-of-care assays and routine coagulation tests, in critically ill patients; a prospective observational study. PLoS One 11: e0151202. [Crossref]
- Jámbor C, Weber CF, Gerhardt K, Dietrich W, Spannagl M, et al. (2009) Whole blood multiple electrode aggregometry is a reliable point-of-care test of aspirin-induced platelet dysfunction. Anest Analg 109: 25-31. [Crossref]
- Karon BS, Tolan NV, Koch CD, Wockenfus AM, Miller RS, et al. (2014) Precision and reliability of 5 platelet function tests in healthy volunteers and donors on daily antiplatelet agent therapy. Clin Chem 60: 1524-1531. [Crossref]
- Can MM, Tanboğa İH, Türkyılmaz E, Karabay CY, Akgun T, et al. (2010) The risk of false results in the assessment of platelet function in the absence of antiplatelet medication: Comparision of the PFA- 100, multiplate electrical impedance aggregometry and verify now assays. Thromb Res 125: e132-e137. [Crossref]
- Zeymer U, Hohlfeld T, Vom Dahl J, Erbel R, Münzel T, et al. (2017) Prospective, randomised trial of the time dependent antiplatelet effects of 500 mg and 250 mg acetylsalicylic acid i. v. and 300 mg p. o. in ACS (ACUTE). Thromb Haemost 117: 625-635. [Crossref]
- Benedek IH, Joshi AS, Pieniaszek HJ, King SY, Kornhauser DM (1995) Variability in the pharmacokinetics and pharmacodynamics of low dose aspirin in healthy male volunteers. J Clin Pharmacol 35: 1181-1186. [Crossref]
- Mannucci PM (2000) Desmopressin (DDAVP) in the treatment of bleeding disorders: the first twenty years. Haemophilia 1: 60-67. [Crossref]
- Svensson PJ, Bergqvist PB, Juul KV, Berntorp E (2014) Desmopressin in treatment of haematological disorders and in prevention of surgical bleeding. Blood Rev 28: 95-102. [Crossref]
- Lethagen S, Olofsson L, Frick K, Berntorp E, Björkman S (2000) Effect kinetics of desmopressin-induced platelet retention in healthy volunteers treated with aspirin or placebo. Haemophilia 6: 15-20. [Crossref]
- Pearson K, Jensen H, Kander T, Schött U (2016) Desmopressin in vitro effects on platelet function, monitored with Multiplate, ROTEM and Sonoclot. Scand J Clin Lab Invest 76: 282-290. [Crossref]
- Berlin G, Hammar M, Tapper L, Tynngård N (2019) Effects of age, gender and menstrual cycle on platelet function assessed by impedance aggregometry. Platelets 30: 473-479. [Crossref]
- Dalby MC, Davidson SJ, Burman JF, Davies SW (2000) Diurnal variation in platelet aggregation with the PFA–100 platelet function analyser. Platelets 11: 320-324. [Crossref]
- Krekels JP, Verhezen PW, Henskens YM (2019) Platelet aggregation in healthy participants is not affected by smoking, drinking coffee, consuming a high-fat meal, or performing physical exercise. Clin Appl Thromb Hemost 25: 1076029618782445. [Crossref]
- Feldman M, Cryer B (1999) Aspirin absorption rates and platelet inhibition times with 325-mg buffered aspirin tablets (chewed or swallowed intact) and with buffered aspirin solution. Am J Cardiol 84: 404-409. [Crossref]
- Cox D, Maree AO, Dooley M, Conroy R, Byrne MF, et al. (2006) Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers. Stroke 37: 2153-2158. [Crossref]
- Diapharma (2015) Multiplate Package Insert Compendium. https://diapharma.com/wp- content/uploads/2015/07/Multiplate_Package_Insert_Compendium_R4.pdf.
- Würtz M, Hvas A-M, Christensen KH, Rubak P, Kristensen SD, et al. (2014) Rapid evaluation of platelet function using the Multiplate® Analyzer. Platelets 25: 628-633. [Crossref]
- Al-Azzam SI, Alzoubi KH, Khabour O, Alowidi A, Tawalbeh D (2012) The prevalence and factors associated with aspirin resistance in patients premedicated with aspirin. Acta Cardiol 67: 445-448. [Crossref]
- von Pape K-W, Dzijan-Horn M, Bohner J, Spannagl M, Weisser H, et al. (2007) Control of aspirin effect in chronic cardiovascular patients using two whole blood platelet function assays: PFA-100 and multiple electrode aggregometry. Hamostaseologie 27: 155-60.
- https://www.fass.se/LIF/product?userType=0&nplId=19920313000031&docType=6&
- Schwertner HA, McGlasson D, Christopher M, Bush AC (2006) Effects of different aspirin formulations on platelet aggregation times and on plasma salicylate concentrations. Thromb Res 118: 529-534. [Crossref]
- Rubak P, Villadsen K, Hvas A-M (2012) Reference intervals for platelet aggregation assessed by multiple electrode platelet aggregometry. Thromb Res 130: 420- 423. [Crossref]
- Peerschke EI, Castellone DD, Stroobants A, Francis J (2014) Reference range determination for whole-blood platelet aggregation using the Multiplate analyzer. Am J Clin Pathol 142: 647-656. [Crossref]
- Consuegra‐Sánchez L, López‐Palop R, Cano P, Carrillo P, Picó F, et al. (2013) Assessment of high on‐treatment platelet reactivity in patients with ischemic heart disease: concordance between the Multiplate and VerifyNow assays. J Thromb Haemost 11: 379-381. [Crossref]
- Neergaard-Petersen S, Ajjan R, Hvas A-M, Hess K, Larsen SB, et al. (2013) Fibrin clot structure and platelet aggregation in patients with aspirin treatment failure. PLoS One 8: e71150.
- Mehta JL, Mohandas B (2010) Aspirin resistance: Fact or fiction? A point of view. World J Cardiol 2: 280-288. [Crossref]
- Helten C, Naguib D, Dannenberg L, Pöhl M, Ayhan A, et al. (2018) Platelet function testing: dead or alive. J Thromb Haemost 16: 984-986.
- Schoergenhofer C, Hobl EL, Schwameis M, Gelbenegger G, Staudinger T, et al. (2017) Acetylsalicylic acid in critically ill patients: a cross‐sectional and a randomized trial. Eur J Clin Invest 47: 504-512. [Crossref]
- Yi X, Cheng W, Lin J, Zhou Q, Wang C (2016) Interaction between COX-1 and COX-2 variants associated with aspirin resistance in chinese stroke patients. J Stroke Cerebrovasc Dis 25: 2136-2144. [Crossref]
- Nagatsuka K, Miyata S, Kada A, Kawamura A, Nakagawara J, et al. (2016) Cardiovascular events occur independently of high on-aspirin platelet reactivity and residual COX-1 activity in stable cardiovascular patients. Thromb Haemost 116: 356-368. [Crossref]
- Taylor ML, Misso NL, Stewart GA, Thompson PJ (1992) The effects of varying doses of aspirin on human platelet activation induced by PAF, collagen and arachidonic acid. Br J Clin Pharmacol 33: 25-31. [Crossref]
- Djarv T, Swain JM, Chang WT, Zideman DA, Singletary E (2020) Early or First Aid Administration Versus Late or In-hospital Administration of Aspirin for Non-traumatic Adult Chest Pain: A Systematic Review. Cureus 12: e6862. [Crossref]
- Pedersen SB, Grove EL, Nielsen HL, Mortensen J, Kristensen SD, et al. (2009) Evaluation of aspirin response by Multiplate® whole blood aggregometry and light transmission aggregometry. Platelets 20: 415-420. [Crossref]
- Desborough MJ, Oakland KA, Landoni G, Crivellari M, Doree C, et al. (2017) Desmopressin for treatment of platelet dysfunction and reversal of antiplatelet agents: a systematic review and meta- analysis of randomized controlled trials. J Thromb Haemost 15: 263-272. [Crossref]
- Powner DJ, Hartwell EA, Hoots WK (2005) Counteracting the effects of anticoagulants and antiplatelet agents during neurosurgical emergencies. Neurosurgery 57: 823-831. [Crossref]
- Gerstein NS, Schulman PM, Gerstein WH, Petersen TR, Tawil I (2012) Should more patients continue aspirin therapy perioperatively?: clinical impact of aspirin withdrawal syndrome. Ann Surg 255: 811-819. [Crossref]
- Weber CF, Dietrich W, Spannagl M, Hofstetter C, Jámbor C (2010) A point-of-care assessment of the effects of desmopressin on impaired platelet function using multiple electrode whole-blood aggregometry in patients after cardiac surgery. Anesth Analg 110: 702-707. [Crossref]
- Mannucci P, Canciani M, Rota L, Donovan BS (1981) Response of factor VIII/von Willebrand factor to DDAVP in healthy subjects and patients with haemophilia A and von Willebrand's disease. Br J Haematol 47: 283-293. [Crossref]
- Fuchs B, Budde U, Schulz A, Kessler CM, Fisseau C, et al. (2010) Flow-based measurements of von Willebrand factor (VWF) function: binding to collagen and platelet adhesion under physiological shear rate. Thromb Res 125: 239-245. [Crossref]