We describe an infant with prenatally diagnosed muscular ventricular septal defect (VSD) and dilated cardiomyopathy with clinical deterioration despite escalating medical therapy in whom successful transcatheter muscular VSD closure was achieved eliminating need to pursue cardiac transplantation.
Dilated Cardiomyopathy is the most common form of pediatric cardiomyopathy, with the majority of patients presenting in the first year of life [1]. Alexander et al. reported a 26% rate of death or transplant within the first year of diagnosis and worse survival among those diagnosed at less than 4 weeks of age [2]. Ventricular septal defects are the most common form of congenital heart disease, reports have described an incidence of 2% to 5% including small muscular defects [3]. Based on the size of the defect and the pulmonary vascular resistance, infants can manifest symptoms of heart failure from pulmonary overcirculation as early as 2 weeks of age. Muscular VSDs are difficult to close surgically due to the right ventricular trabeculations and the difficulty visualizing the defect from the right atrium [4,5]. We present a case of an infant born with dilated cardiomyopathy and a mid-muscular VSD with significant heart failure symptoms poorly responsive to medical therapy. This patient underwent transcatheter VSD device closure at 2 months of age with subsequent clinical improvement and normalization of ejection fraction.
A newborn male was delivered at 39 weeks gestation with an in utero diagnosis of high muscular VSD and biventricular dilated cardiomyopathy. Prior fetal imaging at 23 weeks had demonstrated the VSD with normal biventricular function. Follow up ultrasound at 35 weeks gestation showed reduced biventricular function. There were no signs of hydrops and the biophysical profile was normal. His postnatal echo demonstrated biventricular dilation and moderate to severely decreased ventricular function (biplane ejection fraction, 29%), moderate unrestrictive midmuscular VSD with left-to-right shunting, and small secundum ASD with left-to-right shunting. Newborn screen was normal and urine CMV was negative. His mother was known to have 3.51 Mb deletion on 6q25.4 – 6q26 causing short stature and chromosomal microarray demonstrated this same abnormality for our patient. He was started on heart failure therapy with spironolactone and enalapril given the echo findings of reduced ventricular function and was discharged after 7 days.
Follow up at 3 weeks of age suggested heart failure symptoms with hepatomegaly, a gallop rhythm, and suboptimal weight gain of 10 grams per day, therefore furosemide was added. At 4 weeks of age, he was admitted for 10 days for continued poor weight gain and his medical regimen was escalated to include carvedilol and NG supplementation with formula fortified to 27 kcal/oz. At discharge he weighed 3365 g with a combination of oral and NG feedings providing 120 kcal/kg/day.
His physiology was consistent with a combination of pulmonary overcirculation and left atrial hypertension secondary to the VSD and decreased biventricular function from dilated cardiomyopathy. He was discussed at our surgical conference regarding the elimination of his left-to-right shunt by closure of his VSD and it was felt that transcatheter approach would be preferable to avoid cardiopulmonary bypass given his reduced biventricular function.
At 8 weeks of age and 3.5 kg, he was admitted one day prior to catheterization to initiate milrinone for inotropic support periprocedure. Catheterization was performed using transesophageal echo guidance which demonstrated a 6-7 mm midmuscular ventricular septal defect. Initial closure was attempted with an 8 mm Amplatzer muscular VSD device, however this caused LV outflow tract obstruction so was recaptured prior to release. Following, a 6 mm Amplatzer muscular VSD device was deployed and released and did not cause outflow tract obstruction but partially entrapped the septal leaflet of the tricuspid valve causing tricuspid insufficiency. The case was complicated by transient 2:1 heart block, junctional ectopic tachycardia, and ventricular tachycardia which responded to amiodarone infusion. He was extubated on postprocedure day 3, off inotropes on postprocedure day 5, and transferred out of ICU on postprocedure day 6, with hospital discharge occurring on postprocedure day 12. His discharge medications included: amiodarone, carvedilol, enalapril, spironolactone, furosemide, and enoxaparin.
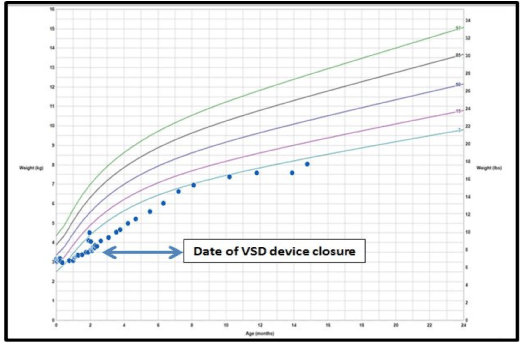
Figure 1. WHO growth chart demonstrating increase in growth velocity following VSD closure
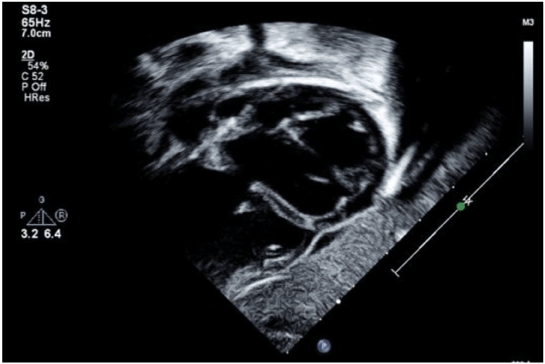
Figure 2. Subxiphoid sagittal image at end-systole demonstrating 5 mm high muscular ventricular septal defect and severe biventricular enlargement. Measured ejection fraction 29%. Image obtained from University of Virginia
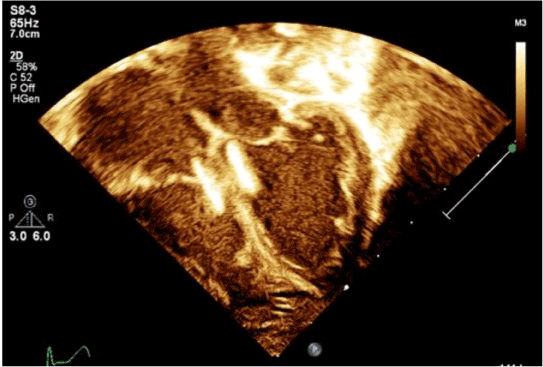
Figure 3. Apical four-chamber view at end-systole on postprocedure day 1 demonstrating position of VSD device in high muscular septum with severe biventricular enlargement. Measured ejection fraction on this study was 23%. Image obtained from University of Virginia
Since discharge, he has shown consistent improvement in clinical status with discontinuation of the NG tube, enoxaparin and amiodarone at 2 weeks, 3 weeks and 6 months post-procedure, respectively. At present, he is 14 months of age and has had no recurrence of dysrhythmia or heart failure symptoms while being medically managed with a typical pediatric heart failure regimen (carvedilol, enalapril, furosemide, and spironolactone). He has no physical exam findings suggestive of congestion. His most recent echo demonstrated mild tricuspid insufficiency predicting RV systolic pressure of 28 mmHg, normal RV systolic function, and low normal LV systolic function with an ejection fraction of 56%.
The most common pediatric cardiomyopathy is dilated cardiomyopathy and most of these patients present in the first year of life [1]. Fetal diagnosis of cardiomyopathy carries a grave prognosis with a high rate of intrauterine fetal demise or death as an infant. Schmidt et al. reported a case series of 6 fetuses diagnosed with cardiomyopathy with death occurring in 1 fetus and 3 infants. Of the two survivors, one underwent heart transplantation at 5 months of age and the other had resolution of symptoms at 2 years of age and was 6 years old at the time of publication [6]. Yinon et al. reported a series of 12 fetuses diagnosed with cardiomyopathy, three of which were terminated. Of the nine remaining, five were complicated by fetal/infant death with the four remaining having normalization of cardiac function by one month of age [7].
Therapeutic strategies include medically optimizing circulation with diuretics, afterload reduction and continuous inotropic support. Invasive measures include reducing metabolic demand with endotracheal intubation/mechanical ventilation and augmenting systemic flow with ventricular assist devices. For those with continued symptoms without recovery of function, cardiac transplantation is offered, though organ availability is limited making wait times variable. Those diagnosed at less than 4 weeks of age have a reported 26% rate of death or need for cardiac transplant [2].
Ventricular septal defects are the most common form of congenital heart disease with an incidence of 2% to 5% [3]. Based on the size of the defect and the pulmonary vascular resistance, infants can manifest symptoms of heart failure from left to right shunt as early as 2 weeks of age. Surgical repair of muscular VSDs is typically challenging due to the heavily trabeculated right ventricle and inability to visualize the defect through a right atrial approach [4,5]. In select cases, transcatheter closure of muscular VSDs has shown promise though the adverse event rates in children weighing less than 5 kg are ~50% [8]. This case illustrates the ability of transcatheter intervention to manage muscular VSD in the setting of fetal cardiomyopathy, thereby avoiding cardiopulmonary bypass and potentially contributing to normalization of myocardial function. There was significant, though mostly transient, morbidity associated with the procedure.
- Lipshultz SE, Sleeper LA, Towbin JA, Lowe AM, Orav EJ, et al. (2003) The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med 348: 1647-1655. [Crossref]
- Alexander PM, Daubeney PE, Nugent AW, Lee KJ, Turner C, et al. (2013) Long-term outcomes of dilated cardiomyopathy diagnosed during childhood: Results from a national population-based study of childhood cardiomyopathy. Circulation 128: 2039-2046. [Crossref]
- Hoffman JI, Kaplan S (2002) The incidence of congenital heart disease. J Am Coll Cardiol 39: 1890-1900. [Crossref]
- McDaniel NL, Gutgesell HP (2008) Ventricular Septal Defects. In: Allen HD, Driscoll DJ, Shaddy RE, Feltes TF (edtrs). Moss and Adams’ Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adults. (7th Edn). Lippincott Williams & Wilkins: 668-682.
- Holzer R, Balzer D, Cao QL, Lock K, Hijazi ZM (2004) Device closure of muscular ventricular septal defects using the Amplatzer muscular ventricular septal defect occluder: Immediate and mid-term results of a U.S. registry. J Am Coll Cardiol 43: 1257-1263. [Crossref]
- Schmidt KG, Birk E, Silverman NH, Scagnelli SA (1989) Echocardiographic evaluation of dilated cardiomyopathy in the human fetus. Am J Cardiol 63: 599-605. [Crossref]
2021 Copyright OAT. All rights reserv
- Yinon Y, Yagel S, Hegesh J, Weisz B, Mazaki-Tovi S, et al. (2007) Fetal cardiomyopathy - in utero evaluation and clinical significance. Prenat Diagn 27: 23-28. [Crossref]
- Lock JE, Block PC, McKay RG, Baim DS, Keane JF (1988) Transcatheter closure of ventricular septal defects. Circulation 78: 361-368. [Crossref]