Treatment of Cystic Fibrosis (CF) is shifting from symptom-based therapies to CFTR-modulating therapies that treat the basis of disease [1]. Potentiators and correctors are the two major classes of CFTR protein modulating drugs, and they are becoming available for increasing numbers of patients [1-4]. Subjects with rare mutations are usually not included in clinical trials and therefore cannot benefit from modulator treatment.
We describe the clinical response of two CF patients who are homozygous to an extra-rare mutation - Q359K/T360. This mutation has so far been described exclusively in Jewish CF patients of Georgian decent [4-6].
The mutation was characterized as a change of C to A at 1207 and at 1211 in exon 8 results in a change of Gln to Lys at position 359, as well as Thr to Lys at position 360 [4].
In an Israeli study, Mei Zahav et al. describe 8 people with at least one allele carrying the Q359K/T360 mutation. All the patients in the cohort had severe pulmonary disease, abnormal sweat test, abnormal nasal potential difference test and sputum culture typical to CF. The authors concluded that the Q359K/T360 mutation is a CF-causing mutation, likely with minimal CFTR function [4]
According to a structural model, the Q359K/T360K mutation may interfere with chloride conductance through steric hindrance and/or through the formation of an “electrostatic trap” [4].
We examined the response to modulators in an organoid culture from one of our patients with the Q359K/T360K mutation.
Organoids are a useful tool for precise quantification of CFTR function in vitro [7-9,14].
In the organoid culture taken from our patient, no significant swelling was seen with VX-770 and/or VX-809 at the predefined forskolin concentration of 0.128mM. With higher concentrations of forskolin, VX-770 causes significant swelling that is further enhanced by the addition of VX-809.
The conclusion was that this patient may benefit from the treatment with Ivacaftor/Lumacaftor (Orkambi).
Following the positive information about the organoid culture test response, the two patients described here were treated with modulators.
Patient 1
The first patient is a 30 year old female, homozygous to the Q359K/T360 mutation. She suffers from pancreatic insufficiency with severe bronchoectatic pulmonary disease. In August, 2018, she began treatment (self-financed) with Ivacaftor/Lumacaftor (figure 1).
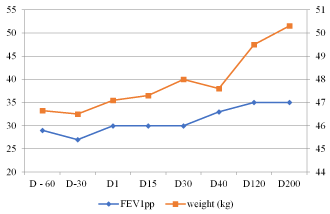
Figure 1. Patient 1 - Change in FEV1pp and weight (kg) with Orkambi treatment
Pulmonary function test: Her FEV1 was 31% predicted (pp) at the beginning of treatment. After 8 months of treatment FEV1 was 35pp (12% improvement).
Weight: Her weight was 46.5 kg.
CFTR function: The patient’s sweat test (conductivity) value was 108 mmol/liter before treatment. After 15 days of treatment sweat test value was 55 mmol/liter (normal result), indicating influence of the drug on CFTR function.
Patient 2
A 36-year-old male, homozygous to the Q359K/T360 mutation. He suffers from bronchoectatic pulmonary disease and pancreatic insufficiency. This patient received funding for one month of Lumacaftor/Ivacaftor (Orkambi) treatment and one month of Tezacaftor/Ivacaftor (Symdeko) treatment. During the first month, the patient was treated with Orkambi. Then he was taken off the drug for two weeks, after which he was treated with Symdeko for another 4 weeks.
Pulmonary function test: Before beginning treatment, his FEV1 was 48pp. Significant improvement in FEV1pp was observed on day 15 of treatment with Orkambi - 35% (to a value of FEV1 65pp). After 14 days without modulator treatment there was an 18% deterioration of FEV1pp (to a value of FEV1 55pp). After 30 days of treatment with Symdeko there was an even more significant improvement of FEV1pp - 53% (to a value of FEV173pp).
Weight: His weight was 63 kg before the treatment, and he gained 5 kg in 10 weeks.
CFTR function: The baseline sweat test value was 86 mmol/liter. On day 15 of treatment with Orkambi, the sweat test value was 45 mmol/liter (-41). After 14 days of washout, sweat conductivity value was an abnormal 107 (+62). After 15 days of Symdeko, sweat test value dropped to a value of 66 (-41) (intermediate result). The effect of Orkambi and Symdeko on sweat conductivity was similar to that observed in patients with the Gly551Asp (12) mutation who were treated with Ivacaftor monotherapy (Figure 2).
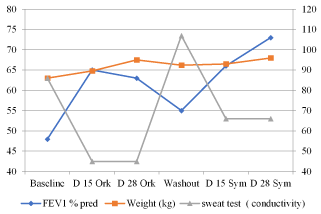
Figure 2. Patient 2 - Change in FEV1 pp, weight(kg) and sweat test conductivity with and without modulator treatment
In this report we describe two CF patients with an extra rare mutation. Following an in vitro assay, we assumed that they would benefit from modulator treatment. Because of the rarity of the mutation, they are not candidates for clinical trials, and we had to settle for collecting clinical data. The results, however, indicate a significant clinical benefit.
FEV1 is a key end point for the evaluation of new therapies for CF and decreased FEV1 is associated with increased risk of death. (1-3,10-11) In the two patients discussed in this report there was a significant improvement of FEV1pp.
Patients with CF typically have difficulty gaining and maintaining weight (1-3.10-11). In this report, the patients gained 3.5 and 5 kg. The results of sweat conductivity indicate the influence on CFTR (1-3 11-13) – improving with modulator treatment and deteriorating without treatment.
In conclusion, CF patients homozygous to Q359K/T360K mutation may benefit from modulator therapy.
No conflict of interest exists with regard to the manuscript.
- Beekman Jeffrey M (2016) Individualized medicine using intestinal responses to CFTR potentiators and correctors. Pediatr pulmonol 51 S44: S23-S34. [Crossref]
- Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, et al. (2017) Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Phe508del and a Residual Function Mutation. N Engl J Med 377: 2024. [Crossref]
- Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, et al. (2019) Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med. [Crossref]
- Mei-Zahav M, Stafler P, Senderowitz H, Bentur L, Livnat G, et al. (2018) The Q359K/T360K mutation causes cystic fibrosis in Georgian Jews. J Cyst Fibros 17: e41-e45 [Crossref]
- Mussaffi H, Prais D, Mei-Zahav M, Blau H (2006) Cystic fibrosis mutations with widely variable phenotype: the D1152H example. Pediatr Pulmonol 41: 250-254. [Crossref]
- Kerem, Eitan, Kalman YM, Yahav Y, Shoshani T, Abeliovich D, et al. (1995) Highly variable incidence of cystic fibrosis and different mutation distribution among different Jewish ethnic groups in Israel. Hum genet 96: 193-197. [Crossref]
- Mou H, Brazauskas K, Rajagopal J (2015) Personalized medicine for cystic fibrosis: establishing human model systems. Pediatr pulmonol 50 S40: S14-23. [Crossref]
- Clancy JP, Jain M (2012) Personalized medicine in cystic fibrosis: dawning of a new era. Am J Respir Crit Care Med 186: 593-597. [Crossref]
- Dekkers JF, Van Mourik P, Vonk AM, Kruisselbrink E, Berkers G, et al. (2016) Potentiator synergy in rectal organoids carrying S1251N, G551D, or F508del CFTR mutations. J Cyst Fibros 15: 568-578. [Crossref]
- Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, et al. (2015) Lumacaftor–Ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 373: 220-231. [Crossref]
- De Boeck K, Munck A, Walker S, Faro A, Hiatt P, et al. (2014) Efficacy and safety of Ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros 13 (2014): 674-680. [Crossref]
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, et al. (2010) Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 363: 1991-2003. [Crossref]
- Eckford PD, Li C, Ramjeesingh M, Bear CE (2012) Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem 287: 36639-36649. [Crossref]
- Chen JM, Cutler C, Jacques C, Boeuf G, Denamur E, et al. (2011) A combined analysis of the cystic fibrosis transmembrane conductance regulator: implications for structure and disease models. Mol Biol Evol 18: 1771-1788. [Crossref]