The Zintl ternary phase, Li18Na2Ge17, have been synthesized typically from a very electropositive metals (Ge) and more electronegative main group metals Na and Li. We attempted to understand its electronic structure and the contribution of electropositive and electronegative groups by employing first-principles calculations. Li18Na2Ge17 is a semiconductor with a narrower and indirect band gap about 0.254 eV. Charge density difference distribution supplemented by Bader population were reported to characterize the bonding. As a ternary Zintl, Li18Na2Ge17 is obeying to the Zintl–Klemm concept, where an electron transfer from the alkali metal atoms (Li, Na) to the clusters (Ge17) is demonstrated.
Zintl phases, Zintl–Klemm concept, DFT, Bader analysis
Zintl clusters have been receiving extensive and lasting interest because of their interesting and diversified structures as well as chemical bonding, unique reactivity, and applications in materials science [1-4]. In fact, Zintl compounds embodying isolated clusters of more than four atoms have been discovered in the early of eighties [3]. These compounds have demonstrated to obey to the Zintl–Klemm concept, known to be “the most important theoretical concept in solid-state chemistry of the last century”, where their structural and bonding characteristics depends on the combination of the electropositive metals (e.g. alkali, alkaline earth and rare earth metals) with the main-group elements (clusters).
The recently synthesized ternary Zintl phase Li18Na2Ge17 [5] has been stabilized by mixing cations of different size and different charge, where a novel structure was obtained with favorable cluster sheathing. The single-crystal X-ray structure has revealed that the ternary Zintl phase Li18Na2Ge17 contains the anionic cluster unit of [Ge12]12− which give to this phase specific features in bonding and electronic properties. In fact, Li18Na2Ge17 have various anions of different sizes, mainly Ge12, Ge4 which need a cation (like Na2 or Li4) to ensure the cohesion of the compound. Cations are important to stabilize these anionic clusters where they can play a role of bridge or separator [6]. The aim of the present paper is to provide a comprehensive picture of the bonding properties and electronic structure of the ternary Li18Na2Ge17. In particular, we addressed the crystal structure, related electronic properties as well as bonding analysis by using ab-initio calculations based on the density functional theory. The electronic structure and chemical bonding investigation have then provided an overall view of electro-structural behavior that can be needed for improving its applications.
Our calculations were performed using the Quantum Wise (ATK) package [7] with the generalized gradient approximation (GGA) of Perdew, Burke and Ernzerhof [8]. ATK code is one of the electronic structure-modeling method as well as software package developed within density functional theory (DFT) by making a use of standard norm-conserving pseudopotential (PP) besides the application of flexible linear numeric combination of atomic orbitals basis set [9]. In these calculations numerical double-ζ plus polarization (DZP) basis set were used [10]. In order to avoid any underestimation in the band gap energy, we used the modified version of the exchange potential proposed by Becke and Johnson (mBJ or meta-GGA) exchange correlation functional [11] and the improved Troullier–Martins norm-conserving pseudopotentials were adopted for all atoms in the Zintl. The structural geometry were optimized by reducing the atomic forces of atoms to be smaller than 0.05 eV/A. The real space grid for the electrostatic potentials is calculated with the mesh cut-off energy of 450 eV which realizes the balance between efficiency and accuracy in the calculation. For Brillouin zone (BZ) sampling, grid of 2 × 2 × 4 k-points for relaxation and structural optimization, and grid of 4 × 4 × 8 k-points for density of states calculation were used. The choice of number of k points and the value of plane wave cutoff energy were considered to ensure the convergence criteria of total energy.
The Li18Na2Ge17 crystallizes in the trigonal space group P31m (No. 157) where the unit cell parameters were found to be a = 17.0905(4) Å, and c = 8.0783(2) Å, with α=β=90 and γ=120 [6] (see Figure1a). Note that the structure contains three different Zintl anions (Z=3). The Li18Na2Ge17 is a ternary of a series of Zintl phases, issued from three different Zintl anions: isolated anions Ge4−, tetrahedra [Ge4]4−, and truncated, Li-centered tetrahedra [Li@Ge12]11−, whose hexagonal faces are capped by four Li cations, resulting in the polyhedra [Li@Li4Ge12]7− as displayed in Figure 1(b-d). Moreover, the structure incorporates other coordination environments as the isolated Ge atom Ge1 is coordinated by nine Li atoms and Na1 (Figure 1e). The Li18Na2Ge17 structure was relaxed at the experimental lattice parameters. Lattice relaxation was studied, but the atomic positions stayed very close to the positions dictated by symmetry and the lattice constant varied by only about 1%, as reported in Table 1. As can be seen from these data, the relaxed Ge4–Ge5 and Ge2–Ge3 distances within the tetrahedra [Ge4]4−, are 2.599 Å and 2.604 Å, respectively. These results are agree well with the experimental values of 2.569 Å and 2.575 Å. Moreover, the optimized equilibrium parameters, a = 17.186 Å, and c = 8.106 Å, were found to corroborate with the experimental data [10]. The deviations from experimental values were overestimated for all quantities by 0.56% and 0.34%, respectively.
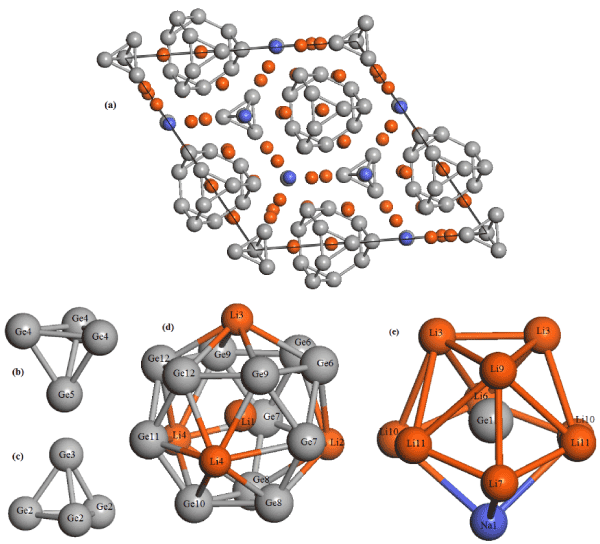
Figure 1. (a) Crystal structure of Li18Na2Ge17 (Li, orange; Na, bleu; Ge, gray),
(b, c) different tetrahedra [Ge4]4−,
(d) Structure of the Li-centered Friauf polyhedron [Li@Li4Ge12]7−, and
(e) the isolated Ge atom Ge1 is coordinated by nine Li atoms and Na1 (Li, orange; Na, bleu; Ge, gray).
Table 1. Selection of distances of new Zintl phase Li18Na2Ge17.
|
Our work |
Exp. |
|
Our work |
Exp. |
Ge1 – Li6 |
2.539 |
2.514 |
Ge7 – Li2 |
3.007 |
2.980 |
Ge1 – Li7 |
2.742 |
2.709 |
Ge7 – Li4 |
2.882 |
2.882 |
Ge1 – Li9 |
2.592 |
2.560 |
Ge8 – Ge8 |
2.734 |
2.695 |
Ge1 – Li11 |
2.569 |
2.543 |
Ge8 – Ge10 |
2.592 |
2.560 |
Ge1 – Li13 |
2.671 |
2.643 |
Ge8 – Li2 |
2.926 |
2.916 |
Ge1 – Li10 |
2.927 |
2.902 |
Ge8 – Li4 |
2.842 |
2.851 |
Ge2 – Ge3 |
2.604 |
2.575 |
Ge9 – Ge12 |
2.522 |
2.500 |
Ge2 – Ge2 |
2.598 |
2.561 |
Ge9 – Li3 |
2.944 |
2.935 |
Ge4 – Ge5 |
2.599 |
2.569 |
Ge9 – Li4 |
3.008 |
2.978 |
Ge4 – Ge4 |
2.627 |
2.582 |
Ge10 – Ge11 |
2.504 |
2.484 |
Ge6 – Ge6 |
2.516 |
2.488 |
Ge11 – Ge12 |
2.619 |
2.592 |
Ge6 – Ge7 |
2.698 |
2.657 |
Ge11 – Li4 |
2.870 |
2.871 |
Ge6 – Ge9 |
2.629 |
2.598 |
Ge12 – Ge12 |
2.609 |
2.599 |
Ge6 – Li2 |
3.001 |
2.974 |
Ge12 – Li3 |
2.881 |
2.857 |
Ge6 – Li3 |
2.921 |
2.906 |
Li1 – Li3 |
2.992 |
2.981 |
Ge7 – Ge8 |
2.532 |
2.495 |
Li1 – Li2 |
2.936 |
2.940 |
Ge7 – Ge9 |
2.596 |
2.565 |
Li1 – Li4 |
2.992 |
3.009 |
To understand the interplay between the electronic and crystal structures, theoretical analysis of the band structure and total/partial density of states (DOS) has been carried out for Li18Na2Ge17 with complete occupation of all positions. As shown in Figure 2 (a, b), the band structure and DOS reveal an indirect narrow band gap (A®G) approximately 0.254 eV above the Fermi level and indicate semiconducting behavior. The partial DOS below the Fermi level consists three well separated regions. The low-energy range (E < −6.5 eV) is composed mainly of the Ge(s) states with minor participation of Li(s), Na(s), Na(p), and Ge(p) states (see Figure 3 (a,b)). The region between ~−4 and -0.6 eV is mainly formed by the Ge(p) states with small contribution of Li(s), and Na(p). Furthermore, strong Ge-s orbital contributions to the DOS also appear between −4 and −0.6 eV below the Fermi level as expected for negatively charged Ge atoms. The bands from −0.5 eV to the Fermi level are mainly composed of Ge 4p and Na 3s orbitals. Further analysis of the projected DOS clearly reveals a hybridization between 4p orbitals of Ge with 2p orbitals of Na (in the range from −0.5 eV to the Fermi), while orbitals of Li do not have much mixing with Ge orbitals.
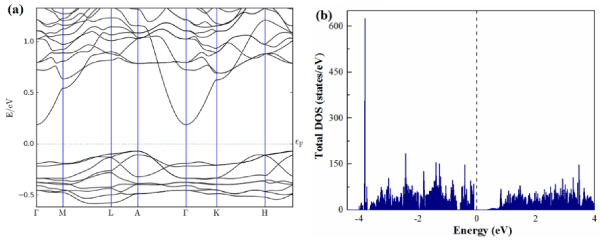
Figure 2. (a) Electronic band structure and (b) total density of states of Li18Na2Ge17.
The vertical solid line denotes the Fermi level.
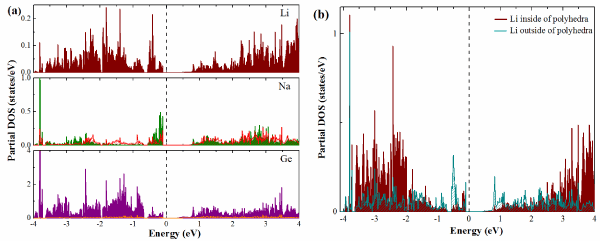
Figure 3. (a) Calculated partial density of states plots for Li18Na2Ge17 compound. Wine, green, red, orange and purple areas represent PDOS of the Li s states, Na s, p states and Ge s, p states, respectively.
(b) Partial projections of orbital components Li 2s inside (wine area) and outside (dark cyan area) of polyhedron [Li@Li4Ge12]7−.
Now we turn to the alkali-metals Na and Li contributions as presented in the electronic structure. The alkali-metals cations was playing an important role in linking or separating different Ge clusters and contributed to stabilize the Zintl anions [6]. In Li18Na2Ge17 structure, the Li atoms occupy two sites: the first position is located in the center of the [Li@Li4Ge12]7− polyhedra with a hexagonal arrangement; the second position is situated outside of the polyhedra. However, we have plotted the PDOS Li s states of each position in Figure 3b. It can be stated that the Li 2s states related to [Li@Li4Ge12]7− polyhedra show a complete electron transfer from the Li (Alkali atom) to the cluster (Ge12) compared to the Li located outside the cluster. This result shows that Li18Na2Ge17 fulfills the concept of Zintl-Klemm [12, 13]. Further insight into the organization of the crystal structures was obtained by applying the Bader analysis of atomic interactions [13]. Bader charge analysis indicates that the isolated Ge atom, coordinated by nine Li atoms and Na (Figure 1e), has acquired more the charges about -2.964 than the Ge tetrahedron -1.198e (Figure 1b,c), and polyhedral -0.841e (Figure 1d). In accordance with the electronegativities of the constituting elements, the Ge species have the largest average negative charge -1.28e, while the Li and Na show a positive charge to be +0.886e and +0.826e, respectively. Obviously, the charge transfer is playing an important role in the organization of the Li18Na2Ge17 crystal structure.
Analysis of the charge density difference reveals maxima of the charge accumulation between Ge atoms and shows a charge especially around the Li-centered Friauf polyhedron [Li@Li4Ge12]7− framework, as shown in figure 4. Thus, Ge12- is completed to [Li@Li4Ge12]7- deltahedral cluster of 17 atoms with covalent bonds [14]. Furthermore, the charge density isosurface is showing that the occupied bonding and nonbonding states are always centered at Ge atoms. These calculations confirm the Bader population analysis and clearly show that the charge is localized in both, the bonding and the lone pair regions [15].
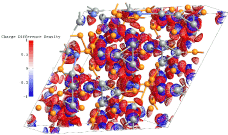
Figure 4. Charge density isosurfaces displaying the bonding character of Li18Na2Ge17.
In summary, we used the state of art of density functional theory to analyze the electronic structure and bonding characteristics of the ternary Zintl phase Li18Na2Ge17. Our calculations have revealed the existence of a balance between packing efficiency and structure, electronic nature, and ions interactions.
Financial support for this study is acknowledged in the form of an internal grants: IRG 18418 from office of research at Alfaisal University.
- Corbett JD (2000) Polyanionic Clusters and Networks of the Early p-Element Metals in the Solid State: Beyond the Zintl Boundary Angew Chem Int. Ed. 39: 670-690.
- Arnold M. Guloy (1996) Chemistry, Structures and Bonding of Zintl Phases and Ions; Kauzlarich, S. M., Ed. VCH: Weinheim, Germany p: 245.
- Von Schnering HG (1981) Angew. Chem. Int. Ed. Engl. 93: 20-33,44.
- Scharfe S, Kraus F, Stegmaier S, Schier A, Fässler TF (2011) Zintl ions, cage compounds, and intermetalloid clusters of Group 14 and Group 15 elements. Angew Chem Int Ed Engl 50: 3630−3670.
- Sevov SC, Goicoechea JM (2006) Chemistry of Deltahedral Zintl Ions. Organometallics 25: 5678−5692.
- Scherf LM, Zeilinger M, Fässler TF (2014) Li18Na2Ge17—A Compound Demonstrating Cation Effects on Cluster Shapes and Crystal Packing in Ternary Zintl Phases. Inorg Chem 53: 2096–2101.
- Soler JM, Artacho E, Gale JD, Garcia A, Junquera J, et al. (2002) The SIESTA method for ab initio order-N materials simulation. D. J. Phys. Condens. Matter 14: 2745–2779.
- Perdew JP, Burke K, Ernzerhof M (1996) Generalized Gradient Approximation Made Simple Phys. Rev. Lett 77: 3865.
- Troullier N, Martins JL (1991) Efficient pseudopotentials for plane-wave calculations. Phys Rev B 43: 1993.
- Junquera J, Paz Ó, Sánchez-Portal D, Artacho E (2001) Numerical atomic orbitals for linear-scaling calculations Phys. Rev. B 64: 235111.
- Tran F, Blaha P (2009) Accurate Band Gaps of Semiconductors and Insulators with a Semilocal Exchange-Correlation Potential Phys. Rev. Lett 102: 226401.
- Bobev S, Sevov SC (2001) Synthesis and Characterization of RbLi7Ge8 with Isolated closo-[Li4Ge12]8- Ions, Lithium-Capped Truncated Tetrahedra of Ge1212-. Angew. Chem. Int. Ed. 40: 8.
- Hoser AA, Jarzembska KN, Dobrzycki Ł, Gutmann MJ, Woźniak K (2012) Differences in Charge Density Distribution and Stability of Two Polymorphs of Benzidine Dihydrochloride. Cryst. Growth Des 12: 3526–3539.
- Bader RFW (1994) Atoms in Molecules, A Quantum Theory; Clarendon Press and Oxford University Press Inc: New York.
- Nesper R (2003) Structural and Electronic Systematics in Zintl Phases of the Tetrels. In: Jutzi P, Schubert U (eds). Silicon Chemistry. Wiley-VCH, Weinheim, 171.
2021 Copyright OAT. All rights reserv