Objective: To describe the mutation frequencies in ataxia genes referred to a clinical laboratory for genetic testing and compare the data with the 2014 European Federation of Neurological Societies (EFNS) testing guidelines.
Methods: Specimens (n=3,394) submitted for hereditary ataxia testing included a 20-gene comprehensive profile (n=2,165), a 14-gene autosomal dominant (AD) profile (n=812), or a 6-gene autosomal recessive (AR) profile (n=417). The genes associated with AD inheritance included ATN1 (DRPLA), ATXN1 (SCA1), ATXN2 (SCA2), ATXN3 (SCA3), SPTBN2 (SCA5), CACNA1A (SCA6), ATXN7 (SCA7), ATXN8OS (SCA8), ATXN10 (SCA10), PPP2R2B (SCA12), KCNC3 (SCA13), PRKCG (SCA14), TBP (SCA17), and AFG3L2 (SCA28). The genes associated with AR inheritance included APTX, FXN [Friedreich ataxia (FRDA)], POLG, SETX, SIL1, and TTPA. Repeat expansions were analyzed by PCR, repeat-primed PCR, and Southern blot assay. Sequence variants were analyzed by Sanger sequencing.
Results: The comprehensive profile yielded positive results for one or more genes in 254 (11.7%) specimens including 204 (80.3%) AD, and 50 (19.7%) AR. Most positive findings (92.9%) were repeat expansions with 7.1% pathogenic variants. Expansions in SCA8 (18.1%) were the most common positive results followed by SCA3 and SCA6 (16.1% each), FRDA (15.7%), SCA2 (13.8%), SCA1 (6.7%), SCA10 (2.4%), SCA17 (2.0%), SCA7 (1.6%), and SCA12 (0.4%). SCA1, 2, 3, 6, 7, and 17 accounted for 81.6% of positive results in the AD profile while FRDA accounted for 78.9% in the AR profile.
Conclusion: The mutation frequencies reported, except for SCA8 and SCA10, are consistent with the EFNS genetic testing recommendations for ataxia.
Hereditary ataxia, spinocerebellar ataxia, Friedreich ataxia, mutation, genetic testing, autosomal dominant, autosomal recessive
The hereditary ataxias are characterized by progressive incoordination that mainly affects ambulation but often involves the upper extremities, speech, and eye movements [1]. Prevalence varies across populations with estimates ranging from 1 to 9 per 100,000 people [2-5]. Inheritance patterns can be autosomal dominant (AD), autosomal recessive (AR), X-linked, or maternal if part of a mitochondrial genetic syndrome [1]. The most frequent inheritance pattern is AD, which is generally associated with an adult onset. The most common spinocerebellar ataxia (SCA) loci 1, 2, 3, 6, and 7 are characterized by CAG trinucleotide repeat expansions in their respective genes [1]. Among the AR ataxias, which usually have a childhood onset, Friedreich ataxia (FRDA) is the most common with a disease frequency of 1:20,000 to 1:50,000 [1].
Diagnosing specific ataxia subtypes has important treatment and prognostic implications. The diagnosis typically requires ataxia, a detailed family history, the identification of additional clinical features, and neuroimaging. Molecular testing plays a prominent diagnostic role because of the difficulty differentiating subtypes based on clinical grounds alone [1]. In 2014, the European Federation of Neurological Societies (EFNS) recommended initial genetic testing for AD ataxia, including SCA1, 2, 3, 6, 7, and 17 and one gene, FXN (FRDA); it recommended testing for AR ataxia when cerebellar atrophy is absent on brain MRI [6, 7]. These seven loci (SCA1, 2, 3, 6, 7, 17, and FRDA) represent the majority of hereditary ataxias in Europe and more than 60% of AD cases worldwide [8]. The EFNS did not include SCA10 or dentatorubral-pallidoluysian atrophy (DRPLA) in their recommendations because pathogenic variants in these genes are restricted to patients with ancestral ties to Latin American or Asian countries [6,7,9]. Although SCA8 accounts for 2% to 5% of AD ataxia, the EFNS did not recommend testing for this subtype in initial screening because the causative expanded allele has a low penetrance [6,7,10].
The EFNS recommendations for molecular diagnosis of ataxia are designed to assist European neurologists in using genetic testing to diagnose specific ataxia subtypes [6,7]. To evaluate the EFNS recommendations for testing specific ataxia genes, we retrospectively assessed the frequencies and types of mutations in specimens referred to a clinical laboratory in the USA.
This study used de-identified laboratory results from consecutive specimens submitted for ataxia genetic testing between 2011 and 2014. Less than 1% of specimens were received from outside the USA. The results from sequencing and repeat expansion assays were extracted from an internal database by gene name without identifying information. In all cases, healthcare providers ordered genetic testing on a standardized requisition form and provided the clinical indications and ICD codes. Detailed clinical information and the relatedness between patients were unavailable.
The study included 3,394 patient specimens, 2,165 of which were tested using a comprehensive profile of 20 genes: 14 associated with AD and 6 associated with AR ataxias (Table 1). The remaining specimens were tested with a 14-gene AD profile (n=812) or a 6-gene AR profile (n=417) (Table 1). Non-repeat expansions were detected by DNA sequencing, which was performed by PCR amplification of purified genomic DNA, followed by Sanger DNA sequencing of the coding regions of all the genes except POLG and KCNC3. POLG gene exons 7, 13, and 21 and KCNC3 gene exon 2 were sequenced and analyzed. Abnormal sequence variants were confirmed using bi-directional sequencing. Repeat expansions were detected by PCR amplification of the repeat region followed by high-resolution electrophoresis to determine the number of tandem repeats in each allele. Repeat primed assay or Southern blot analysis was used to confirm the homozygosity of normal alleles. A two-tailed Fisher’s exact test was used to compare the differences in the positive rates between the EFNS [7] and the clinical laboratory datasets.
Table 1. Ataxia genes (loci) included in multigene profiles tested in a US clinical laboratory
Comprehensive Profile |
Dominant Profile |
Recessive Profile |
ATN1 (DRPLA) |
ATN1 (DRPLA) |
APTX |
AFG3L2 (SCA28) |
AFG3L2 (SCA28) |
FXN (FRDA) |
APTX |
ATXN1 (SCA1) |
POLG |
ATXN1 (SCA1) |
ATXN2 (SCA2) |
SETX |
ATXN2 (SCA2) |
ATXN3 (SCA3) |
SIL1 |
ATXN3 (SCA3) |
ATXN7 (SCA7) |
TTPA |
ATXN7 (SCA7) |
ATXN8OS (SCA8) |
|
ATXN8OS (SCA8) |
CACNA1A (SCA6) |
|
CACNA1A (SCA6) |
ATXN10 (SCA10) |
|
ATXN10 (SCA10) |
KCNC3 (SCA13) |
|
FXN (FRDA) |
PPP2RB2 (SCA12) |
|
KCNC3 (SCA13) |
PRKCG (SCA14) |
|
POLG |
SPTBN2 (SCA5) |
|
PPP2RB2 (SCA12) |
TBP (SCA17) |
|
PRKCG (SCA14) |
|
|
SETX |
|
|
SIL1 |
|
|
SPTBN2 (SCA5) |
|
|
TBP (SCA17) |
|
|
TTPA |
|
|
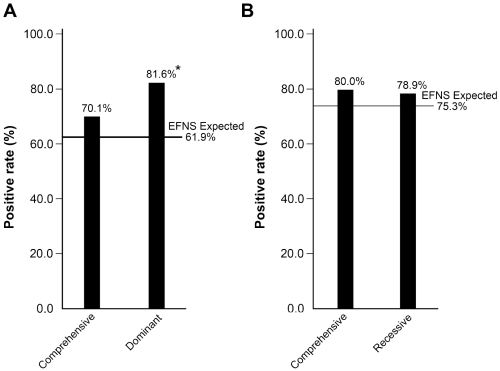
Of the 2,165 patients tested with the comprehensive profile, 254 (11.7%) were positive for repeat expansions, pathogenic variants, or likely pathogenic variants: 204 (80.3%) in AD and 50 (19.7%) in AR ataxia genes. Repeat expansions accounted for 92.9% (236/254) of the positive results, and pathogenic or likely pathogenic variants for only 7.1% (18/254). The positive rates by gene for the comprehensive profiles are shown in Figure 1A. Repeat expansions in genes associated with AD ataxia collectively accounted for 196 (77.2%) of the 254 positive results. SCA8 expansions were the most frequent accounting for 18.1% of the 254 positive results, followed by SCA3 (16.1%), SCA6 (16.1%), SCA2 (13.8%), SCA1 (6.7%), SCA10 (2.4%), SCA17 (2.0%), SCA7 (1.6%), and SCA12 (0.4%); no DRPLA mutations were detected (Figure 1A). There were no differences in the regional geographic distribution of positive results, except for SCA10 (n=24): positive results clustered in California (14/24) and Texas (3/24). Repeat expansions in FXN (FRDA) accounted for 15.7% of positive results (Figure 1A). The positive rates for both the AD profile (20.1%, 163/812) (Figure 1B) and AR profile (4.6%, 19/417) (Figure 1C) were higher than those for genes within the comprehensive profile. For the comprehensive profile, the positive rates were 9.4% (204/2,165) for genes associated with AD and 2.3% (50/2,165) for genes associated with AR ataxia. The 6 AD genes recommended by the EFNS, ATXN1 (SCA1), ATXN2 (SCA2), ATXN3 (SCA3), CACNA1A (SCA6), ATXN7 (SCA7), and TBP (SCA17) accounted for 70.1% (n=143) of the positive results for AD ataxia genes (n=204) in the comprehensive profile (Figure 2A). These frequencies were not significantly different (P=0.095) than the frequencies reported by the EFNS (61.9%) [7]. However, these 6 genes accounted for 81.6% in the AD-only profile which is significantly more than expected (P<0.001) compared to the EFNS data (Figure 2A) [7]. The observed frequencies of positive results for FRDA (80.0%) in the comprehensive AR profile and in the AR-only profile (78.9%) were not significantly (P=0.810) different from the EFNS frequency (75.3%) reported for the European population (Figure 2B).
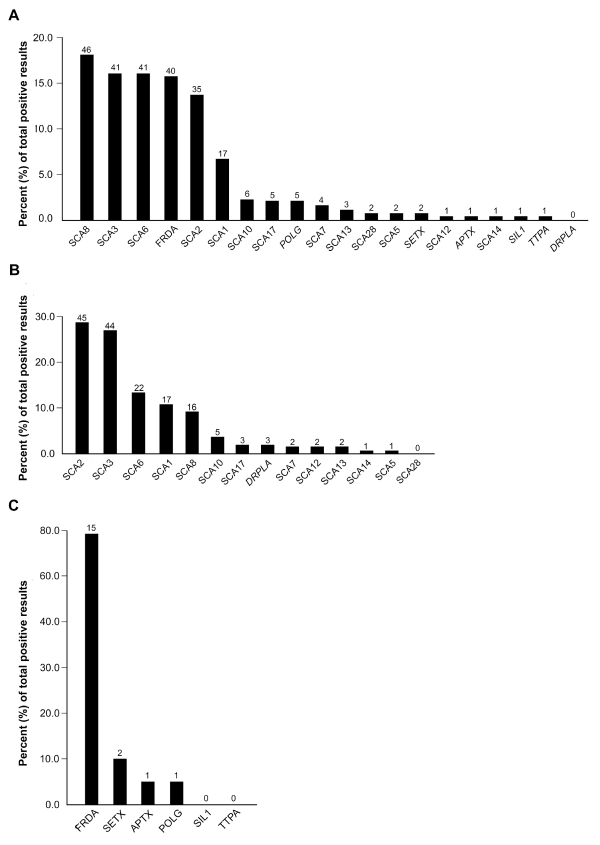
Figure 1. The frequency of positive test results for loci or genes in the comprehensive (A), AD (B), or AR (C) profiles (n=436). The percent of pathogenic or likely pathogenic test results for each profile are shown. The numbers above the black bars are the numbers of positive results in the total numbers of positive tests for the comprehensive (n=254), AD (n=163) and AR profiles (n=19).
2021 Copyright OAT. All rights reserv
Figure 2. The frequencies of positive results for the (A) dominant loci (SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17) and (B) recessive locus (FRDA) as compared to the observed frequencies published by the European Federation of Neurological Societies (EFNS) in 2014 (horizontal lines) [7]. The frequencies of positive results in the comprehensive and AR profiles were similar to the EFNS data. The frequency of positive results for the AD profile was significantly (P<0.001) higher compared to the EFNS data.
The positive rates in this study may be overestimates due to the lack of detailed clinical information and the possibility that more than one family member was tested. Overall, the data agrees with the 2014 EFNS guidelines for genetic testing for ataxia with two exceptions [6, 7]. The first exception is that SCA10 is not included in the EFNS recommendation because it has been only been reported in individuals from Latin American or of Native American descent [9]. However, in our study population, SCA10 accounted for 2.4% of our positive specimens; we noted a regional clustering in California and Texas likely due to founder effects. The second exception is that repeat expansions in SCA8 were the most common positive result (18.1%) in the comprehensive profile. The high rate of co-occurrence with pathogenic variants in other AD ataxia genes in this study (10/46 specimens: 4 SCA8 expansions on both alleles, 4 with other repeat expansions, and 2 with a sequencing variant in POLG) suggest that repeat expansions in SCA8 may not cause ataxia directly in some cases. In addition, not all SCA8 expansions are associated with progressive ataxia suggesting the co-occurrence is due to its low penetrance [8,10]. The role of SCA8 as a genetic modifier and the mechanisms for incomplete penetrance require further study.
The comprehensive profile detected AD mutations in nearly 10% of ataxia patients and AR mutations in only 2.3% in our heterogeneous sample. When an AD or AR profile was ordered, the frequency of positive results roughly doubled. This suggests that the AD-only or AR-only profiles should be performed first in patients with a known inheritance pattern.
This study largely supports the EFNS recommendations for genetic testing for ataxia, except for the exclusion of SCA10. The inclusion of SCA8 testing remains controversial due to incomplete penetrance and the finding of co-occurrence with other ataxia gene mutations. Multigene profiles for pathogenic variants in known ataxia genes are especially useful when the inheritance pattern is known.
- Jayadev S, Bird TD (2013) Hereditary ataxias: overview. Genet Med 15: 673-683. [Crossref]
- Durr A (2010) Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 9: 885-894. [Crossref]
- Joo BE, Lee CN, Park KW (2012) Prevalence rate and functional status of cerebellar ataxia in Korea. Cerebellum 11: 733-738. [Crossref]
- Coutinho P, Ruano L, Loureiro JL, Cruz VT, Barros J, et al. (2013) Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 70: 746-755. [Crossref]
- Koht J, Tallaksen CM (2007) Cerebellar ataxia in the eastern and southern parts of Norway. Acta Neurol Scand Suppl 187: 76-79. [Crossref]
- Gasser T, Finsterer J, Baets J, Van Broeckhoven C, Di Donato S, et al. (2010) EFNS guidelines on the molecular diagnosis of ataxias and spastic paraplegias. Eur J Neurol 17: 179-188. [Crossref]
- van de Warrenburg BP, van Gaalen J, Boesch S, Burgunder JM, Dürr A, et al. (2014) EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood. Eur J Neurol 21: 552-562. [Crossref]
- Paulson HL (2009) The spinocerebellar ataxias. J Neuroophthalmol 29: 227-237. [Crossref]
- Bushara K, Bower M, Liu J, Karen N McFarland, Ivette Landrian, et al. (2013) Expansion of the Spinocerebellar ataxia type 10 (SCA10) repeat in a patient with Sioux Native American ancestry. PLoS One 8: e81342. [Crossref]
- Zeman A, Stone J, Porteous M, Burns E, Barron L, et al. (2004) Spinocerebellar ataxia type 8 in Scotland: genetic and clinical features in seven unrelated cases and a review of published reports. J Neurol Neurosurg Psychiatry 75: 459-465. [Crossref]