Abstract
From an aetiological standpoint, viral cardiomyopathy represents an uncommon subtype of non-inflammatory dilated cardiomyopathy. The most common aetiologic agents are enteroviruses, adenoviruses and erythroviruses. Pathogenesis depends on the causative virus. Enteroviruses and adenoviruses infect and injure the cardiomyocyte through cytopathic effect and immune-mediated damage leading to cardiac remodelling, myocarditis and ultimately cardiomyopathy. Erythroviruses infect and injure the vascular endothelial cells resulting in macrovascular dysfunction. Typical clinical presentation is heart failure, arrhythmias and chest pains. Clinical diagnosis requires the presence of electrocardiographic abnormalities, markers of myocardial necrosis or evidence of functional/structural ventricular abnormalities accompanied by at least one physical sign or clinical symptom. However, endomyocardial biopsy remains the reference standard but increased risks of complications and the need for highly experienced operators limits its widespread use. The available clinical management strategies are standard heart failure medication for the management of cardiac dysfunction and antiarrhythmic drugs for those with ventricular arrhythmias. Patients with refractory symptoms greater than six months despite optimal medical therapy and with biopsy-proven virus negative myocardium may benefit from supplementary immunosuppressive therapy. However, large-scale and long-term prospective randomized clinical trials are warranted to determine long-term benefits of immunosuppression.
Key words
dilated cardiomyopathy, viral cardiomyopathy, viral myocarditis
Introduction
Classification systems in clinical medicine have been pivotal in facilitating the development of standardized disease nomenclature, focused disease research and the development of safe and efficacious clinical management strategies. In the case of cardiomyopathies (CM), classification has proved exceedingly complex. Underpinning this complexity is considerable phenotypic overlap and heterogeneous clinical presentations between categories and in the same category during the natural course of the disease. The World Health Organization (WHO) and the European Society of Cardiology (ESC) propose a morphofunctional classification while the American Heart Association (AHA) propose primary vs secondary classification based on myocardial and/or organ involvement [1-4]. All these classifications suffer significant overlap between individual categories. Aetiological classification is also imperfect since categories with similar genotypes may exhibit different phenotypes and pathogenic pathways and vice-versa [3]. Nevertheless, the most recent MOGES classification that incorporates morphofunctional, organ-involvement, inheritance pattern, aetiology and underlying disease underscores the importance of aetiological classification in advancing the knowledge and understanding of the pathogenesis of a disease [5,6]. This paper provides a review of available published evidence and expert consensus on virus aetiologies of CM including the role of aetiologies in the diagnosis and treatment of viral CM.
Clinical definitions
The 2006 AHA scientific statement on contemporary definitions and classification of the cardiomyopathies defines CM as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic” (pp. 1809) [4]. The 2008 ESC position statement on classification of the cardiomyopathies defines CM as “a myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease and congenital heart disease sufficient to cause the observed myocardial abnormality” (p. 271) [2]. Dilated cardiomyopathy (DCM) in which virus forms part of the aetiologic agents, is a phenotype characterized by ventricular dilation and depressed myocardial performance in the absence of coronary artery disease or abnormal loading conditions [3]. Thus, viral CM may be considered a sub-type of DCM defined by viral persistence in a dilated heart. When viral persistence is accompanied by myocardial inflammation, the disease may be termed inflammatory CM or viral myocarditis (VMC) with cardiomegaly. However, if there is no biopsy evidence of inflammation on a dilated heart (< 14 lymphocytes and macrophages/mm²), then the term viral CM (VCM) or viral persistence DCM should be applied [7].
Epidemiology
Viral infection of the heart is relatively rare and usually asymptomatic with spontaneous and complete resolution. However, in uncommon cases, it may lead to substantial cardiac damage, the development of VMC, VCM and congestive heart failure (HF) [7]. Usually, VMC occurs in all age groups from infants to older adults but it is prevalent in children and adults under the age of 40, with 35% of the patients aged 10-30 years old [8]. However, accurate determination of the prevalence and incidence of viral heart infection has been problematic due to a wide variety of viruses and several periods of epidemic, which lead to significant differences in the predominant viruses in different regions as well as in different years within the same region. In addition, the low use of virological tests has resulted in few epidemiological data on viral heart infection. However, three categories of epidemiological data provide important insights into the prevalence of viral heart infection: (a) data from autopsy or biopsy examinations; (c) data from clinical diagnosis during periods of viral epidemic; and (c) data from population-based studies.
Data from autopsy/biopsy examination
Available autopsy and biopsy data suggest a very low prevalence of both viral heart infection and VMC. Analysis of 377,841 cases of autopsy between 1958 and 1977 from the Japanese Pathology Society found low incidence of non-specific myocarditis (0.11%) and tuberculoid myocarditis (0.007%) [9]. In Italy, an analysis of 17,162 autopsy cases between 1965 and 1994 reported a low incidence of VMC (0.53%) [10]. The European Study of Epidemiology and Treatment of Cardiac Inflammatory Diseases (ESTCID) conducted between 1993 and 1999 investigating endomyocardial biopsies of 3,055 patients found 526 cases (17.2%) of acute or chronic VMC [11].
Data from clinical diagnosis
Data from clinical diagnosis of viral infection of the heart during viral epidemic periods indicate significantly higher incidence of between 5 and 10%. In 1981 during the period of influenza epidemic in China, virus antibodies were positive in 43% of 183 patients with fever, in which 13 cases were consistent with clinical diagnosis of VMC with an incidence of 7.1% [11]. Paired virus serum antibody was positive in 41% of 1,426 VMC-suspected patients between 1978 and 1986, which was similar to the year 1981, where reported cases of VMC was 28% in 393 patients with incidence increasing up to 28% [7,12].
Data from population-based studies
Population-based studies report very low incidence of viral heart infection and myocarditis. A collaborative group in nine provinces and cities in China between 1978 and 1980 investigating the incidence of VMC reported 1,709 paediatric, 136 VMC-suspected, and 90 cardiomyopathy, with VMC incidence ranging between 6.8 to 29.2 per 100,000 [4]. A review of 1,349,828 deaths in Finland between 1970 and 1998 reported an incidence of 0.47 per 1,000 deaths due to myocarditis. The incidence remained constant in the 1970s through to the 1980s rising in the 1990s [13]. In Yunnan province in China, variation in prevalence was associated with income levels and geographic locations, with an average incidence of 1.2% between 1978 and 2004 [14]. At present, the incidence of VMC in China has risen from the 10th to the 4th leading heart disease based on records of patients hospitalized with heart diseases [15].
Aetiologic Agents
A broad spectrum of infectious agents is involved in the pathogenesis of viral CM. The spectrum varies with geographical region, patient’s age, treatment used and the presence of concomitant diseases [7]. Table 1 provides a list of the common aetiologic agents of VCM alongside their primary or main diseases. Although numerous viruses may be involved in the initial myocardial infection and subsequent development of VCM, the most commonly observed viruses in VCM patients are erythrovirus (Parvovirus B19), enteroviruses (coxsackievirus) and adenoviruses [16]. However, it is almost possible to quantify the exact frequency that cardiomyotropic viral infection lead to clinically significant VMC and VCM. Such quantification would require tissue sampling from otherwise healthy individuals during a viral epidemic [7].
Table 1. Aetiologic Agents of Viral Myocarditis and Other Associated Diseases
Aetiologic Agents |
Common Diseases Associated with Infection |
1. Coxsackievirus |
Hand, foot and mouth disease, herpangina or pleurodynia (Bornholm disease) [17-31]. |
2. Parvovirus |
Erythema infectiosum (fifth disease), polyarthropathy, transient aplastic crisis, pure red cell aplasia, hydrops fetalis or congenital anaemia [32-34]. |
3. Adenovirus |
Keratoconjunctivitis, respiratory and enteric infection gastroenteritis, hepatitis, pneumonia, meningoencephalitis, cystitis, upper or lower respiratory tract infections [35-40]. |
4. Herpes virus |
Exanthema subitum (sixth disease), encephalitis, mesial temporal lobe epilepsy and multiple sclerosis [41-48] |
5. Cytomegalovirus |
CMV mononucleosis or CMV-associated graft failure [50-56]. |
6. Varicella virus |
Chicken pox, herpes zoster (shingles) [57-62]. |
7. Hepatitis virus |
Acute/chronic hepatitis or hepatocellular carcinoma [63-70] |
8. Influenza virus |
Influenza [71-77] |
9. Poliovirus |
Poliomyelitis [78-83] |
10. Mumps virus |
Mumps [84-86] |
11. Rubella virus |
Rubella (German measles), congenital rubella syndrome [87,89] |
12. Rubeola virus |
Measles [90-94] |
13. Variola |
Smallpox [95-98] |
14. Epstein-Barr |
Infectious mononucleosis, epithelial and lymphocytic carcinoma [99-105] |
15. Echovirus |
Aseptic meningitis, encephalitis [106-110] |
16. Rabies virus |
Rabies [111-116] |
17. Mycoplasma virus |
Viral pneumonia [117-122] |
18. Psittacosis virus |
Atypical pneumonia (psittacosis) [123-126] |
19. HIV |
Acquired Immunodeficiency Virus (AIDS) [127-139] |
20. Arbovirus |
Encephalitis, epidemic mosquito-borne arboviruses (yellow fever
virus, dengue virus, West Nile virus, chikungunya virus and Zika virus) [140-144] |
Coxsackievirus
Coxsackievirus is a member of the picornaviridae family in the enterovirus genus of viruses. They are positive-sense single-stranded RNA viruses divided into coxsackievirus A (CVA) and B (CVB) species [17]. The CVA species is the major causative agent of both epidemic and sporadic cases of hand, foot and mouth disease, and herpangina (painful mouth blisters) [18-20]. In children, CVB is a unique cause of syndromes such as myopericarditis and pleurodynia (Bornholm disease). Other CVB-related diseases include infections of the central nervous system, respiratory tract and vertically transmitted infections (mother-to-child/embryo) [21]. Coxsackievirus infections are also responsible for several inflammatory conditions including myocarditis, pericarditis, pancreatitis, meningitis and encephalitis [22]. Coxsackievirus is a common cause of acute MC in children or young adults (< 35 years) [23-25] and about a half of healthy individuals have detectable serum antibodies indicating prior infection [26-28]. Tests based on polymerase chain reaction (PCR) reveal positive-strand enteroviral RNA in 35% of DCM patients [29]. Analysis of a German registry data shows low incidence of enteroviral MC (3%) and enteroviral CM with or without inflammation (4% each) [7]. Necropsy analysis of enteroviral CM patients reveal pericardial effusion, cardiomegaly, and a predominant mononuclear inflammatory infiltrate accompanied by necrosis of the atrial and ventricular myocardium, and in some patients, focal myocardial necrosis mimicking myocardial infarction despite normal coronary arteries [30]. Cardiac susceptibility to viral infection is due to affinity for myocardial membrane receptors (human Coxsackie-adenovirus receptor [hCAR]) to viral particles [31].
Parvovirus
Parvovirus B19 (PVB19) is a member of the Parvoviridae family in the erythrovirus genus of small round, non-enveloped single-stranded RNA virus [32]. It is an autonomously replicating virus and the main site of infection is erythrocyte precursors [33]. The PVB19 virus is widespread and the clinical picture associated with its infection vary based on the immunologic and hematologic status of the infected individual. In healthy immunocompetent children, PVB19 is the aetiologic agent of erythema infectiosum (fifth disease), an innocuous rash illness. In adults, infection is occasionally associated with an acute symmetric polyarthropathy mimicking rheumatoid arthritis. Due to tropism of PVB19 to erythroid progenitor cells, infection in individuals with an underlying haemolytic disease causes transient aplastic crisis. In immunocompromised individual, persistent PVB19 infection may cause pure red cell aplasia and chronic anaemia. Infection in foetus may lead to death in utero, hydrops fetalis or congenital anaemia [32,33]. In rare cases, PVB19 infection has been associated with several syndromes including vasculitis, encephalitis, pruritis, congenital red cell aplasia, chronic bone marrow failure and Kawasaki disease [33]. Since the heart and kidney express receptors for PVB19, it could lead to myocarditis, and complications and/or rejection in liver and renal transplant patients [33]. Recently, PVB19 infection has been associated with myocarditis and viral cardiomyopathy with high mean numbers of virus copies in EMB – 2013 in inflammatory DCM compared to 57 in non-inflammatory DCM and 44 in HCM [7]. Pankuweit et al. analysis of PCR series reported up to 30% of endomyocardial biopsy (EMB) samples in patients with DCM and MC [34].
Adenovirus
Human adenovirus (HAdV) is a non-enveloped, double stranded DNA virus belonging to the family Adenoviridae in the genus Mastadenovirus that contains seven known species: HAdV-A to HAdV-G [35-37]. The primary sites of infection include the gastrointestinal tract, lung, urinary tract, upper respiratory tract and eye [35]. Common transmission pathways include exposure to infected individuals via inhalation of contaminated aerosolized droplets or direct conjunctival inoculation, or through faecal-oral spread such as contact with infected recreational fresh-water or tap water, airflow filters or environmental surfaces [38]. Although HAdV are prevalent in water bodies such as in rivers, coastal waters, swimming pool and drinking water, they can retain their infectious properties for several weeks in moisture free environments, and are resistance to disinfectants [38,39]. HAdV infection mainly causes keratoconjunctivitis as well as have been associated with complications such as gastroenteritis, hepatitis, myocarditis and pneumonia mostly in children (< 5 years) [36]. HAdV infection accounts for 3% to 5% of acute respiratory infections in children and < 2% in civilian adults [40]. In immunocompromised patients, infection has been associated with high morbidity and mortality [37-39]. Relative to other viral causes of CM, it is the second most frequent virus (after Coxsackievirus) found by PCR examinations in EMB of patients with viral cardiomyopathy [40]. In patients with MC and DCM, positive PCR ranges between 5% and 8% [7].
Human herpes virus
Human herpes virus (HHV-6) is a double stranded DNA virus belonging to Betaherpesvirinae subfamily in the genus Roseolovirus with two closely related yet distinct variants: HHV-6A and HHV-6B, which are two of the eight herpes virus (HHV1-8) in which the human body is the primary host [41-43]. Over 95% of the individuals older than two years are seropositive for HHV-6A and/or HHV-6B. [41]. HHV-6 exhibits a wide cell tropism in vivo (lymph nodes, macrophages and monocytes, kidney tubule endothelial cells, salivary glands and CNS tissues, and induces a lifelong latent infection [41,42]. In immunocompromised individuals (by either natural means or pharmacologic interventions), HHV-6 may cause serious disease including exanthema subitum (sixth disease: a benign disease of infancy) as a primary infection while further virus reactivation can induce severe encephalitis mostly in hematopoietic stem cell transplant patients [42-44]. Due to high tropism for CNS cells, HHV-6 has been associated with a diverse array of neurologic diseases including seizures, encephalitis, mesial temporal lobe epilepsy and multiple sclerosis [43]. Immunocompromised patients such as those undergoing renal/bone marrow transplant are at a greater risk for post-transplant disease or complications [41]. HHV-6 is a common cause of viral MC (10.5%) and viral CM (21.6%) [45,46]. Although the exact pathogenic role of HHV-6 is still a matter of research, viral persistence or presence may be responsible for fatal MC in children aged between 4 and 24 months or the progression to DCM in infected individuals [47-49].
Cytomegalovirus
Human cytomegalovirus (CMV) is an enveloped double-stranded DNA virus belonging to the viral family Herpesviridae of the genus Cytomegalovirus [50,51]. The CMV can infect any organ but more commonly occur in the blood, brain, colon, heart, kidney, lung and stomach [52]. Primary infection is rare in individuals aged younger than 35 years but prevalent in immunosuppressed individuals [53-55]. Its main transmission route is contact with the mucous membrane or parenterally via blood components containing cells or via stem cell or organ transplant [50]. Other important transmission routes include peri-natal and post-natal mother-to-foetus through transplacental, cervical or vaginal secretions and breast milk, and sexual transmission via cervical secretion and semen or vial the saliva [51]. Immunocompromised patients such as those receiving organ transplants or treatment for HIV/AIDS or cancer are at increased risk of serious complications [50]. Mostly, CMV infection is associated CMV mononucleosis, as well as a host of end-organ diseases (pneumonia, gastrointestinal disease, hepatitis disease, CNS disease, retinitis, nephritis, cystitis, myocarditis, pancreatitis splenomegaly and colitis) and CMV-associated graft failure [50,51]. Cardiac involvement in CMV infection is rare. In a German hospital register, CMV-associated MC was found in <3% of the respective patient cohort [7]. Reported prevalence of CMV in MC and DCM patients is 3% and 0.8% [45,46]. Cardiac infection in adults present as asymptomatic and transient electrocardiographic abnormalities. Symptomatic cardiac involvement is rare but in some immunosuppressed individuals, haemorrhagic pericardial effusion or MC with LV dysfunction, and attendant congestive HF may manifest [54-56].
Varicella Virus
Varicella virus (also known as human herpes virus-3) is a ubiquitous enveloped, linear double stranded DNA virus belonging to the human alpha-herpesvirus family of viruses [57,58]. The virus only naturally infects humans with no animal reservoir. Its main infection targets are T lymphocytes, epithelial cells and ganglia [58]. It spreads by airborne route and its transmission is through the respiratory tract [58,59]. Primary varicella infection causes chicken pox, a common childhood illness associated with fever and general pruritic vesicular rash. Varicella virus establishes latency in the cells of the dorsal root ganglia (even for decades) after the primary infection. It is a less common infection in tropical areas, but in temperate climates, children acquire chicken pox between the fifth and tenth years of life. Second episode of chicken pox is very rare. Reactivation of varicella in the host results in shingles (or herpes zoster), a localized, painful vesicular rash involving one or adjacent dermatomes, whose incidence increases with age or immunosuppression [57-59]. Shingles may be complicated by chronic pain (postherpetic neuralgia) or the presence of other serious neurological and ocular disorders such as meningoencephalitis, myelitis, cranial nerve palsies, vasculopathy, keratitis and retinopathy [58]. MC is an uncommon but a serious complication of varicella infection but unsuspected MC is a common finding in fatal varicella infection [60]. Histological findings may reveal characteristics intranuclear inclusion bodies with myocardial cells accompanied with interstitial oedema, cellular infiltrates and myonecrosis [61]. Varicella infection causing viral MC can progress rapidly to DCM as well as result in life-threatening arrhythmias [60-62].
Hepatitis C Virus
Hepatitis C virus (HCV) is a hepatotropic small enveloped, positive sense, single-stranded RNA virus in the Flaviviridae family and genus hepacivirus [63,64]. The main route of transmission is parenteral exposure via blood or blood components, and a majority of intravenous drug users may become infected by repetitive exposure to contaminated injection equipment [65]. The virus is responsible for 15% to 20% of cases of acute hepatitis. After acute infection, about 50% to 80% develop chronic infection [64]. A subset of patients with chronic hepatitis are at increased risk of developing fatal complications such as cirrhosis (20%) and hepatocellular carcinoma (4%-5%) [63-65]. The involvement of HCV in extrahepatic manifestations such as insulin resistance, Type 2 diabetes mellitus, glomerulopathies and oral manifestations have been described in epidemiological studies [64]. Although cardiac involvement in HCV infection is rare, contested data implicates the virus as an aetiologic agent in some cases of viral CM. [66]. Fulminant MC with congestive HF, hypotension and death may be a consequence of HVC infection but very rare [67,68]. A recent study of 48 non-ischemic DCM patients and depressed systolic function chronic HCV found 4.8% had serum antibodies to the HVC and 2.4% with HCV RNA [69]. Characteristics pathologic features associated with HVC infection include minute foci of necrosis of isolated muscle bundles usually surrounded by lymphocytes and diffuse serious inflammation [70]. The ventricles may also be dilated with petechial haemorrhages [68,70].
Influenza Virus
Influenza virus is an enveloped single stranded, negative-sense, helically shaped RNA virus. The virus belongs to the Orthomyxoviridae family and classified into three distinct types (A, B and C) but only A and B has human latency reservoir [71,72]. Transmission pathway include direct contact with infected nasal discharges, contact with fomites (contaminated objects such as towels or hairbrush) and inhalation of virus-laden aerosols. It is the primary aetiological agent of sporadic and epidemic cases of influenza disease [73]. Cardiac involvement and clinical MC are uncommon in people infected with influenza. However, pre-existing cardiovascular diseases significantly increases the risk of morbidity and mortality in infected patients [74]. In periods of influenza epidemic, about 5% to 10% of infected patients may exhibit symptoms of cardiac involvement [75]. Autopsy findings in fatal cases show biventricular dilatation with mononuclear infiltrate particularly in perivascular area [76]. Although the prevalence of influenza A and B IgG antibodies is high in DCM patients, positive PCR findings in EMB is rare (< 0.5%) [7,77].
Poliovirus
Poliovirus is a member of Enterovirus C, in the family of Picornaviridae. It is a single-stranded positive sense RNA genome [78]. Transmission route of poliovirus is via oral contact with secretion or faecal material from the infected person. Most infection cause asymptomatic viral replication limited to the alimentary canal. Gastrointestinal tract, including the pharyngeal mucosa, is both the portal of entry and primary locus of infection, and the source of viral dissemination [79]. Infection causes poliomyelitis disease, a paralytic disease but 99% of the disease has since been eradicated following the introduction of the polio vaccine [80]. Myocardial involvement after poliovirus infection is very rare, reported in studies in the 1950s with almost no case reports on myocarditis in poliomyelitis in recent years, possible due to successful efforts in eradication of poliomyelitis [81-83].
Mumps Virus
Mumps virus is a member of the Paramyxoviridae family of enveloped, non-segmented, negative-sense RNA viruses. Humans are its only natural host. The virus is highly neurotropic, with evidence of CNS infection in approximately half of cases [84]. The virus causes mumps – a contagious disease that spreads from person to person through respiratory secretions. Before routine vaccination, 95% of adults has serological markers of exposure, which dramatically reduced after the introduction of vaccination. Characteristics of mumps disease are painful swelling of the parotid glands, but can also involve numerous other tissues and organs, resulting in inflammatory reactions including encephalitis, meningitis, orchids, myocarditis, pancreatitis and nephritis [84]. Only about 10% of cases exhibit complications with the involvement of other organs including the heart. Cardiac involvement is a rare complication since the development of mumps vaccine and when observed is described as either pericarditis or acute endocarditis [85]. Myocarditis is rare but a known complication of mumps, with earlier reported incidence of 4-15% [86]. Myocarditis occurs in the first week of the disease and ECG abnormalities usually disappear few weeks later [86].
Rubella Virus
Rubella virus belongs to the Togaviridae family and the only members of the Rubivirus genus of enveloped, positive sense, single stranded RNA virus. Its main transmission pathway is respiratory aerosols to the nasopharyngeal infection and transplacental mother-to-foetus infection. The main site of viral infection and dissemination is the upper respiratory tract and nasopharyngeal lymphoid tissue [87]. Infection by rubella virus causes rubella disease (German measles) and congenital rubella syndrome or miscarriage if rubella infection occurs during the first trimester [88]. Complications of rubella virus include arthralgia or arthritis, encephalitis and haemorrhagic manifestations as well as orchitis, neuritis, and late syndrome of progressive panencephalitis [87]. The incidence of myocardial involvement in post-rubella infection is extremely limited. Reported cases suggest neonatal rubella myocarditis is common due to teratogenicity of rubella virus for developing organs of the human embryo [89].
Rubeola Virus
Rubeola virus belongs to the Paramyxoviridiae family and of the Morbillivirus genus of an enveloped, single-strand, non-segmented negative sense RNA virus. Humans are the only reservoir and has a single serotype [90]. It is highly communicable and spreads through aerosols, direct contact with nasal or throat secretions and less frequently by contact with contaminated surfaces. Once inhaled and a primary target cell (respiratory epithelial cell) is infected, systemic spread ensues and clinical signs appear after 9 to 19 days. Primary infection by rubeola virus causes measles, which is more acute in infants and older people than in children, and rarely causes death [90,91]. Infection confers life-long immunity where second attacks are described as errors in the diagnosis of either the first or the second illness [90]. In rare cases, severe measles-associated CNS complications may occur – acute disseminated encephalomyelitis, measles inclusion body encephalitis or subacute sclerosing panencephalitis [91,92]. Data on the incidence of cardiac complications following measles is extremely limited. Myocarditis and heart block may be common presentation of cardiac abnormalities in measles [93,94].
Variola Virus
Variola virus belongs to the Poxviridae family of the Orthopoxvirus genus of enveloped, non-segmented linear double stranded DNA viruses [95]. The common human exposure routes are inhalation of aerosol through close contact, from fomites or contact with infectious materials in scabs [96]. Infection by variola virus causes smallpox, acute, self-limited human illness with no known human or non-human reservoir. However, the disease was eradicated from the human community towards the end of the 29th Century using vaccinations but mechanisms responsible for the emergence of new dangerous pathogens remain unknown [95,96]. Smallpox-associated cardiac involvement is lacking. However, the use of variola vaccines has been related with serious sequelae mostly myocarditis and pericarditis [97]. Of 450,000 military personnel in the use received variola virus vaccination between 2002 and 2003, two confirmed and 50 probable cases of vaccination-related myocarditis were reported (1.16%) [98]. The majority of cases of myocarditis resolve completely but some patients may develop chronic heart failure and even death [97].
Epstein-Barr Virus
The Epstein-Barr virus, also known as human herpes virus 4, is a gamma-herpes virus that infects the majority of the world’s population [99]. It is one of the most successful viruses infecting more than 90% of humans with lifelong latency. Infection often occurs by contact with oral secretions. The primary site of infection and dissemination is epithelial cells in the oropharynx and almost all seropositive individuals actively shed virus in the saliva [100]. Infection with Epstein-Barr virus is often asymptomatic but may lead to a range of pathologies including infectious mononucleosis to severe cancers of epithelial and lymphocytic origin. Epstein-Barr virus has been detected in tissues from patients with nasopharyngeal carcinoma as well as associated with non-Hodgkin’s lymphoma and oral hairy leucoplakia [100-101]. Besides acute infection accompanied but high fever, a few reports indicate infection by Epstein-Barr virus may lead to fatal outcomes involving myocarditis and sudden cardiac death [102-104]. Epstein-Barr associated myocarditis may also be the first clinical manifestation of infectious mononucleosis [105].
Echovirus
Echovirus belongs to the species Enterovirus B, genus Enterovirus of the Picornaviridae family of small non-enveloped, single-stranded RNA virus, and the largest enterovirus sub-group consisting of 30 serotypes [106-107]. Echoviral infection in humans occurs via faecal-oral transmission and the primary point of infection and dissemination is the nasopharynx to regional lymph nodes. However, infection at non-mucosal site have been reported in a review of literature published before 1985, which reported 61 cases of neonatal echovirus infection [108]. Primary echovirus infection may cause aseptic meningitis and meningoencephalitis [107]. While myocardial involvement in enteroviruses infection are common, echoviruses associated MC occur mostly in childhood and rare in adults. Echovirus MC in adults may be moderate and transient [109]. In immunocompromised children due to leukaemia and immunosuppressive therapy, echovirus infection may lead to transient echovirus MC, with complete symptom resolution within one and a half months [110].
Rabies Virus
Rabies virus (or lyssavirus) is a rod-shaped, single stranded, negative sense enveloped RNA virus belonging to the Rhabdoviridae family of the genus Lyssavirus. It is a neurotropic virus, which causes rabies in both humans and animals [111]. The main transmission pathway of rabies in humans is rabid animal bite, and less commonly via aerosol exposure in laboratory spread and natural settings and organ and tissues transplants. After inoculation, rabies virus enters the peripheral nervous system directly and disseminate to the brain or replicate in muscle tissues prior to CNS invasion and replication [111,112]. At present, no treatment is effective to save the life of a symptomatic rabies patient [112]. Cases of human rabies with associated MC very rare with very few cases reports of the disease between 1960s and 1980s [113]. Focal interstitial MC in which mononuclear cells predominant have been reported in 10 of 23 fatal cases of human rabies [114]. Cheetham et al. [115] reported two cases of rabies MC in the England found at necropsy previously unrecognised but may play a role in the disease, while one case in Zambia reported signs of MC and fever disappeared within 48 hours [116].
Mycoplasma Virus
Little data exists on mycoplasma virus. Mycoplasma are a group of microorganisms of the class Mollicutes, previously known as pleuropneumonia-like organisms. Mollicutes have been shown to carry DNA viruses [117,118]. Morphologically they occur as rods, polyhedrons with short or long tails and as enveloped spheres resembling non-lytic animal viruses and now placed in the new family of bacterial viruses, the Plasmaviridae [118]. Mycoplasma viruses play a more significant pathogenic role in viral diseases than was previously realized, which were observed in a majority of HIV-infected patients suspected to play a synergistic role [119]. Mycoplasma virus causes pneumonia mostly in schoolchildren usually as a co-infection with bacteria [120]. There is very limited data on cardiac involvement following mycoplasma viral infection. Case reports suggest symptomless and transient manifestation in children [121,122].
Psittacosis Virus
Studies and case reports of psittacosis virus are extremely rare with a majority of the available literature dating as far back as the 1950s. Early studies based on serological tests identified two strain of the psittacosis virus: the first originating from pigeons designated as pigeon ornithosis, and the second of an unknown origin [123]. Psittacosis is a latent infection of psittacine birds transmissible to humans causing atypical pneumonia, characterized by high fever stimulating a typhoidal state and symptoms of an atypical pneumonia [124,125]. Rarely, the disease may be transmitted from infected human to another [124]. Cases of myocarditis associated with psittacosis virus us rare and reported cases involve a combination of psittacosis virus and bacteria [126].
human immunodeficiency Virus
Infection with the human immunodeficiency virus (HIV) causes acquired immunodeficiency syndrome (AIDS), manifesting as a profound immunosuppression due to predominant selective depletion of helper/induce T lymphocyte expressing the receptor for the virus (the CD4 molecule) [127]. HIV infection has a high tropism for the brain resulting in neuropsychiatric abnormalities. In addition to inducing cell apoptosis, HIV infection may interfere with T4 cell function. HIV may exist in a latent or chronic form that may be converted to an active infection by a variety of inductive signals [127]. In HIV-infected individuals, cardiac involvement occurs in about 25 to 50%, with clinical heart disease in about 10% [128-136]. Congestive HF with LV dilatation and dysfunction is a common occurrence [137,138]. Endomyocardial biopsy (EMB) evaluation of specimen from 83 HIV-DCM and 80 idiopathic DCM patients reveal a greater mean intensity of tumour necrosis factor - alpha (TNF-α: 0.81) and inducible nitric oxide synthetase (iNOS: 1.007) staining compared to idiopathic DCM (0.44 and 0.49 respectively). The staining intensity of TNF-α and iNOS had an inverse correlation with CD4 count. Staining intensity of iNOS was higher in HIV-DCM patients with HIV/CVB3 or HIV/CMV, which correlated with mortality rate [139].
Arbovirus
Arboviruses belong to the family of viruses transmitted by arthropods (insect vectors) or spread as zoonoses). In the U.S., arboviruses that cause human encephalitis are members of the Togaviridae, Flaviviridae, and Bunyaviridae families [140,141]. Mosquitoes are the main vectors but other biting flies, midges and ticks may also transmit the disease. Humans are incidental hosts who do not produce significant viremia and do not contribute to the transmission cycle. Humans acquire infection during blood feeding by an infected arthropod although laboratory acquired infections may also occur after handling tissues and body fluids [140]. Besides encephalitis, epidemic mosquito-borne arboviruses include yellow fever, dengue, West Nile, chikungunya and Zika virus [140,141]. At present, cardiac complications associated with arbovirus infection is rare. However, case reports in the 1970s describe myocarditis, pericarditis and cardiomyopathy associated with dengue and chikungunya viruses [142,143]. In the 1960s and 70s, dengue-associated myopericarditis was common in Ceylon with ECG abnormalities but with no clinical involvement of the heart [143]. Dengue associated myocarditis has favourable prognosis with resolution of symptoms, improvement in ECG and no residual cardiomegaly but in a few cases may lead to persistence symptoms, cardiomegaly and ECG abnormality transiting to cardiomyopathy [142]. Recently, Zika virus outbreak has been reported in the U.S. and a first travel acquired Zika acute infection complicated in myocarditis to mainland France, which recommends ECG and troponin assessment if any cardiac symptoms are present in a patient with acute Zika infection [144].
Pathogenesis of Cardiac Damage
Damage to the cardiomyocyte
Despite the availability of a well-characterized experimental models and common acceptance that viral infection causes myocarditis, the exact mechanisms underlying pathogenesis in humans remain controversial. At present, the only available evidence comes from animal models of enteroviral MC, which demonstrate a tri-phasic pathogenic process: (a) direct virus-mediated damage; (b) immune-mediated damage; and (c) myocardial remodelling and cardiomyopathy [145-151].
Phase one: Viral-mediated damage
Cardiac involvement after viral infection begins when viruses invade the cardiomyocytes or macrophages through binding with specific receptors and co-receptors. The receptor for CVB and adenovirus 2 and 5 is the hCAR (a junctional protein), which has been supported by the observation that the absence or low abundance of hCAR prevents viral invasion of the cardiomyocytes [152,153]. A co-receptor playing a role in viral invasion for serotypes B1, B2 and B5 is the CVB co-receptor decay-accelerating factor (DAF) [154]. Differential binding to this receptor influences viral virulence [155]. Other determinants of the virulence of CVB include variation in viral genome, and host factors such as selenium deficiency and mercury exposure [156-159]. To obtain a complete understanding of viral-mediated damage to the myocardium, future studies should investigate genetic and environmental determinants of virulence to understand why a great majority of cardiotropic viruses including enteroviruses, adenovirus and parvovirus do not cause cardiomyopathy.
The initial acute phase of active viral replication lasts for a few days in which direct viral damage of the cardiomyocytes occur via virus-mediated lysis [160]. The virus first degrades cell structures to enable entry and damage to the cardiomyocyte [161]. When this damage occurs in the complete absence of host immune, viral proliferation alone is sufficient to initiate severe acute MC with resultant DCM and HF [160]. The innate immune response is the first line of defence against the initial viral invasion. Viral particles and certain host proteins triggers an innate immune response, which involves toll-like receptors (TLRs) and pattern recognition receptors in patients with tissue injury [162]. These receptors recognize foreign antigens and trigger the activation of nuclear transcription factors leading to the production of inflammatory cytokines [163]. In response to massive cytokine production, natural killer (NK) cells and macrophages migrate to the heart and minimalize viral propagation mostly through direct cytotoxic effect [164]. In clinical practice, this initial phase is often asymptomatic because innate immune response prevents initial myocardial damage by eliminating the virus and renovating the damaged tissues [165]. However, in immunocompromised patients, the acute phase of viral replication may present with fever, weakness, rash, muscle pain and joint pain. It may also be accompanied by symptoms of respiratory or gastrointestinal viral infection [150].
Phase two: Viral mediated immune damage
In immunocompetent patients, immune response stimulated by certain host proteins limits viral replication, and in the majority of patients, eliminates the virus from the host. However, immune response itself can cause myocardial damage. In this case, the balance between beneficial and detrimental effects of immune response significantly influences the extent of myocardial cell loss [151]. The characteristics of this second (sub-acute) phase of the pathogenesis of viral CM is viral mediated immune damage to the myocardium, which may last for weeks to months.
This phase is characterized by the production of antibodies (T- and B-lymphocytes), which peak at 7-14 days after virus inoculation, which represents the most severe phase of myocardial damage [151]. These antibodies produced to destroy viral particles may also react with cardiac structure to cause damage to the myocardium. The cytotoxic T-lymphocytes response is the most important mechanism responsible for lysis of virus-infected cells as well as responsible for auto-immune-mediated myocardial damage [150]. The cytotoxic T-cells may attack virally infected cardiomyocytes because of molecular mimicry – myocardial antigens that bear similarities to viral proteins may cause T-cells originally aimed at the virus to cross-react with host antigen and produce myocyte damage. The products of cardiac cell destruction themselves may then stimulate further lysis by T-cells [166]. Murine models have also suggested host-depending genetic factors may increase the risk of autoimmune reactions [148].
A combination of direct viral injury, cytokine context and the level of pro-inflammatory immune reaction determine the severity of MC and the possibility of viral infection shifting from acute to sub-acute phases [164]. The activation of acquired immunity results in chronic inflammatory response in the myocardium and may lead to organ dysfunction due to fibrosis and myocardial remodelling. It may also cause damage due to necrosis and switching on the autophagy of the cardiomyocytes. Although viral genome has been detected in cardiac tissues of patients with chronic MC, the mechanism of long-term persistence of CVB in the presence of intact immune system remains unknown [164,167]. In clinical practice, patients in this second phase may present with dyspnoea, chest pain, heart palpitations, increased exercise intolerance, increased sweating and fainting [148].
Phase three: Cardiac remodelling and myopathy
The third phase of the pathogenesis of viral CM that involves cardiac remodelling and myopathy has been the most difficult to elucidate. The controversy of whether the present of persistent viral genomes in the myocardium or remnants of previous infection contribute to the progression to CM remains unclear. In this third phase, it is usually not possible to detect the virus in the myocardium. In the case of persistence inflammatory response, the heart may develop idiopathic DCM due to myocardial remodelling [148]. Antibodies acting on the sarcolemma, myeloma, beta-receptors, acetylcholine receptor, Laminin and cardiac conduction tissue may play a pathogenic role but the participation of antibodies against fibrils, stress proteins and intermediate filament remains unclear [168]. Inflammation may be followed by the release of cytokines (the transforming growth factor) and the activation of metalloproteinase. Patients with VMC exhibit over expression of the Matrix metalloproteinase (MMPs), which can degrade different components of the cardiac tissue and contribute to myocardial remodelling [169]. Other complementary mechanisms that may play a role in post-inflammatory myocardial remodelling include enhanced fibrosis, which is a consequence of osteopontin and matricellular protein Cyr61 activity [170]. Post-inflammatory cardiac remodelling and myopathy may present with LV systolic dysfunction and LV wall hyperkinesia correlating with ECG abnormalities such as ST-S changes. RV dysfunction is rare. Formation of thrombi within cardiac cavities and pericardial effusion can also occur [148].
Autoantibodies cross-reactivity due to antigen mimicry is another proposed pathogenic mechanism contributing to myocardial remodelling. In patients with lymphocytic MC or DCM, autoantibodies to a variety of antigens is a common histological finding [171,172]. The virus CVB shares epitomes with cardiac myosin (which is an intracellular antigen) and cross-reactive antibodies may lead to the production of autoantibodies due to this antigen mimicry [173,174]. Thus, following virus clearance from the myocardium, cardiac myosin may provide an endogenous source of antigen in chronic MC and stimulate chronic inflammation through autoimmune mechanisms. Antone et al. and Huber et al. have described cross-reactivity between cardiac myosin and endogenous human cell-surface protein Laminin to suggest that Laminin may serve as an ongoing stimulus in chronic antibodies to cardiac myosin cross-reaction with beta-adrenergic receptor, which may contribute to cardiomyocyte apoptosis [172,175,176]. However, the main challenge has been distinguishing antibody autoreactivity occurring in the course of normal immune reaction from that of autoimmune disease in which anti-cardiac antibodies contribute to ongoing CM.
Histologically determined viral presence or viral components in the myocardium independent of local immune response may also lead to fibrosis, hypertrophy and the degeneration of cardiomyocytes, which is observed in DCM patients [177]. Protein products of the enteroviral genome can cleave host proteins, including dystrophin, leading to CM [178]. This induction of dystrophin deficiency augments CM that accompanies the enterovirus [179]. Experimental models indicate CVB might persist in the myocardium with partially deleted genome leading to low-grade non-cytolytic chronic cardiac infection [180]. These findings, if replicated in human DCM patients, might be useful to understand how enterovirus infection can cause chronic DCM in the absence of MC [181]. Pathological changes in the myocardium may provoke chest pain, tachycardia, irregular heartbeat, dyspnoea at rest and during exertion, swelling of lower limbs, fainting and hyperhidrosis [148].
Damage to the vascular endothelial cells
In contrast to enteroviruses and adenoviruses, which infect and injure cardiomyocytes, other common cardiotropic viruses in VMC and VCM such as erythroviruses (parvovirus - HPVB19 and HPVB19V) or human herpesvirus (CMV and HHV6) infect the vascular endothelial cells (ECs) [182-186]. Biopsy samples show parvovirus PVB19 is localized in the ECs of venuoles, small arteries or arterioles in patients with fulminant MC or acute onset of HF [182] and in the ECs of small capillaries in patients with chronic inflammatory CM [183,184]. The primary erythroviruses infection occur predominantly in childhood and the virus persists in the bone marrow of healthy individuals with no recognized clinical significance.
Infection and replication of parvovirus B19 infection is restricted to the erythroid progenitor cells but may also affect the ECs through the distribution of the primary erythroviral receptor (P antigen), and co-receptors like the integrin α5β1 and the KU80 protein [187-189]. The pathogenic mechanisms of ECs damage are complex and may involve cytotoxicity of the non-structural protein 1, transactivation of interleukin-6 (IL-6), and TNF-α and induction of apoptosis [190-195]. Recent evidence suggests the release of parvovirus B19 from the bone marrow occurs through infected capillary precursor cells while interferon-beta (IFN-β) improves viability of B19V infected human ECs [186]. Treatment by IFN-β improves endothelial dysfunction and respective symptoms while both remain unchanged in non-treated patients to suggest a partial role of direct virus-cell interaction mediating B19 induced ECs damage [196].
HHV-6 on the other hand is a lymphotropic virus with tropism mainly for CD4+ and CD8+, B-cell and NK cells but can also infect the vascular ECs [197,198]. HHV-6 specific DNA has been observed in the vascular ECs both in vivo and in vitro to suggest the virus damages the EC [198-200]. It has been postulated that ECs and cardiac myocytes may be important reservoirs for viral latency and re-activation [198]. For HHV-6 and other herpes virus, frequent activation occurs mainly through infections or drugs with sub-acute clinical presentations particularly in acquired or drug-induced immune-deficiencies such as transplant recipients or patients with autoimmune disorders. Mostly, cardiac infection by the HHV-6 enhances the pathogenicity of other viruses more than being a pathogen itself [196]. Viral infection in the ECs may lead to a worsening clinical picture because of microvascular dysfunction, which may predict long-term disease progression in chronic HF [201-204]. Parvoviruses and herpesviruses may also cause cardiac damage through infecting interstitial tissue resulting into heart dilatation and LV dysfunction, and ultimately HF [148].
Clinical Manifestation and Investigations
Clinical presentation
Viral cardiomyopathy has a wide spectrum of clinical presentation ranging from asymptomatic course of a mildly ongoing disease with slightly impaired myocardium to a severe fulminant HF accompanied by malignant arrhythmias. Presentation also varies from one phase of the disease to another. Sudden cardiac death (SCD) may also be the first clinical presentation of the disease in previously healthy individuals [196]. The subclinical nature of the disease during the period of acute viral infection does not exclude a possible evolution of inflammatory myocardial damage while mild symptoms do not indicate favourable long-term prognosis. The extent of myocardial damage in the acute phase may be a determining factor for the recovery of LV function in subsequent phases. Despite the wide heterogeneity, clinical presentation of patients with viral CM often include HF, chest pain and arrhythmias [205-207].
Heart failure
Heart failure is the most common clinical manifestation of viral CM. Although the onset is usually gradual with mild symptoms, in the case of fulminant MC, the development of HF is acute resulting in cardiogenic shock requiring mechanical circulatory support (device therapy) or cardiac transplantation to save the patient’s life. Patients who survive the acute phase show a significant improvement or complete normalization of LV systolic function with favourable long-term prognosis in a few weeks [165]. Thus, the decision for long-term treatment options such as cardioverter, defibrillator, resynchronization or cardiac transplant should be considered after the lapse of the acute phase.
Chest pain
New onset or worsening chest pain either at rest or during exertional activities is an important clinical presentation in patients with viral heart infection. Patients with viral infection of the endothelium may have a higher incidence or worsening chest pain as a manifestation of acute viral heart disease [112]. Usually, chest pain mimics angina pectoris or has pericarditis-like character in the presence of perimyocarditis [58].
Arrhythmias
Arrhythmias is another common complaint and a reason for a visit to a physician. In patients with viral heart disease, arrhythmias may present as supraventricular or ventricular arrhythmias, conduction disturbances or serious ventricular arrhythmias suggesting the possibility of giant cell myocarditis or cardiac sarcoidosis. Symptoms may be present in one patient simultaneously or manifest at different phases of the disease. Symptoms of HF meeting the criteria of inflammatory CM indicate an unfavourable prognosis compared to symptoms of chest pain and arrhythmias [148].
Clinical investigations
Clinical diagnosis of viral CM relies on the current evidence and expert consensus guidelines for the diagnosis of MC, which is a common consequence of acute viral heart infection. Diagnosis of MC is per exclusionem. The AHA statement on diagnosis of forms of DCM and the ESC guidelines on myocardial and pericardial disease
recommend that MC should be suspected in the presence of (a) acute chest pain; new onset or worsening dyspnoea [3,205]; (b) palpitations or unexplained arrhythmias; and (c) unexplained cardiogenic shock. Diagnostic criteria for MC is a combination of clinical findings: ECG abnormalities, the presence of myocardiocytolysis markers, functional/structural abnormalities and tissue characterization by cardiac magnetic resonance imaging (Table 2). The AHA and ESC recommend that clinical diagnosis of MC should be suspected in the presence of the following clinical presentation and/or diagnostic criteria [3,205].
- ≥ 1 clinical presentation and ≥ 1 diagnostic criterion
- ≥ 2 diagnostic criteria, if the patient is asymptomatic
Table 2. Clinical Presentation and Diagnostic Criteria for Viral Myocarditis
Clinical Presentation |
Acute chest pain, pericarditis or pseudoischemic |
New onset (days to 3 months)or worsening dyspnoea at rest or during exertion and/or fatigue with/without sings of left/right HF |
Subacute/chronic (> 3months) or worsening of dyspnoea at rest or during exertion and/or fatigue with/without sings of left/right HF |
Palpitation and/or unexplained arrhythmia symptoms and/or syncope and/or aborted sudden cardiac death. |
Unexplained cardiogenic shock |
Diagnostic Criteria |
ECG abnormalities (AV block, BBB, ST/T-waves changes, supraventricular/ventricular arrhythmias, low voltage QRS complex and abnormal Q waves) |
Markers of myocardial necrosis (elevated cardiac troponins or CK-MB) |
Functional/structural abnormalities in echocardiography or MRI (impaired LV/RV function with/without LV/RV dilatation, increased ventricle wall thickness, pericardial effusion and intracardiac thrombi) |
Tissue characteristic by CMR (presence of at least 2 of 3 Lake Louise criteria, myocardial oedema and early and late gadolinium enhancement) |
AV: atrioventricular; BBB: Bundle Branch Block; ECG: Electrocardiogram; MRI: Magnetic Resonance Imaging
Adapted from AHA Scientific Statement on Diagnosis and Management of Specific DCM [3]
Blood tests
Currently, clinically available and specific blood tests are non-specific and cannot confirm the diagnosis of MC. Serum markers of inflammation such as erythrocyte sedimentation rate and C-reactive protein may be elevated in MC but are non-specific. Biomarkers of cardiac injury such as levels of troponin 1 may be elevated especially in patients with acute and clinically severe MC who require hospitalization but current evidence is inconsistent. In a clinical trial in the U.S. involving subjects with histologically acute MC with an average of one-month symptoms, only 34% of the patients had elevated levels of troponin 1 [3, 205,208]. However, the current troponin assays such as soluble ST2 (a clinically available serum biomarker that also reflects inflammation) promise to be more useful in the diagnosis of MC. The ESC working group position statement recommends troponin levels should be obtained in the clinical setting of suspected MC patients, while the AHA position statement recommends assessment of natriuretic peptides and other biomarkers [3,205].
Electrocardiography
Electrocardiogram (ECG) is a simple conventional examination useful for the initial evaluation of diseases in cardiology. The common ECG abnormality in viral CM are non-specific T-wave changes [209]. Sometimes, the ECG abnormalities may mimic acute myocardial infarction or pericarditis with ST segment elevation, ST segment depression, PR segment depression and pathological Q-waves [210-211]. Often, tachyarrhythmias are non-sustained and rarely result in haemodynamic compromise in adult patients with viral CM. While the prognostic significance and optimal management of non-sustained ventricular tachycardia in acute MC remain unclear, Q-waves and widened QRS complex including left bundle branch block may indicate poor prognosis associated with higher rates of death or the need for cardiac transplantation [209,212-214].
Echocardiography
Echocardiography is a useful low-cost non-invasive imaging modality for excluding other causes of HF as well as for detecting ventricular thrombi. However, echocardiography has no pathognomonic features to use for the diagnosis of patients with viral MC [215,216]. Segmental wall abnormalities may mimic those of myocardial infarction [217]. Patients with fulminant MC may present with a more normal chamber dimension and thickened walls compared to patients with less acute MV who exhibit a greater LV dilation and normal wall thickness [218]. LV dysfunction is rare but an important predictor of death or heart transplantation [219]. New echocardiography imaging techniques such as strain echo may have better specificity for MC [196].
1.1.1 Magnetic Resonance Imaging
Cardiovascular magnetic resonance imaging (CMRI) is useful in patients suspected with MC to localize and quantify the extent of tissue injury including oedema, hyperaemia and fibrosis [220]. Recently, CMRI alone has been shown to confirm correct diagnosis in 80% of a series of 82 patients with biopsy-proven MC [221]. However, diagnosis requires both T1 and T2-weighted imaging to achieve optimal sensitivity and specificity. In contrast to older evidence, CMRI abnormalities do not correlate closely with EMB evidence of MC [204]. When ≥ 2 Lake Louise criteria are positive, myocardial inflammation may be predicted with a diagnostic accuracy of 78%, which drops to 68% with the use of delayed post-gadolinium enhancement [196]. Additional prospective clinical studies evaluating prognostic values of CMRI are needed to determine whether tissue characterization may complement management or outcome of MC patients [220].
Endomyocardial Biopsy
Confirmatory diagnosis of MC still requires histological or immunological evidence of inflammation in cardiac tissues. When performed by highly experienced operators, EMB can have very low major rate of complications, and LV biopsy is as safe as RV biopsy [204,222]. However, there are differences in the indications for EMB by the 2013 AHA/ACCF/ESC joint statement and the 2013 ESC guidelines on HF management working group [205, 223]. The ESC position supports broad use of EMB for the diagnosis and management of MC based on the presence or absence of viral genomes and inflammation. The AHA/ACCF/ESC statement recommends performance of EMB in two group of patients. (1) In patients with HF and normal/dilated LV, > 2 weeks of symptoms and haemodynamic compromise. (2) In patients with dilated ventricle, 2 weeks to 3 months symptoms, new ventricular arrhythmias or Mobitz type II second-degree heart block or those who fail to respond to usual care within 1 to 2 weeks of treatment [223,224]. EMB-based criteria – presence of inflammation by immunohistology and viral genomes absent by PCR – have been used to define a cohort of patients with chronic DCM who responded to immunosuppression therapy [225]. Despite the diagnostic value of EBM, current recommendations support its use only in clinical scenarios where the incremental prognostic and therapeutic information gained outweighs the risk and cost, which varies by medical centre depending on availability of necessary facilities and expertise [205,223].
Clinical management
The pathogenic mechanisms of cardiomyocyte destruction are direct viral damage, anti-viral immune response or autoimmune injury [145-151]. In adults, cardiomyocytes rarely regenerate and recovery of myocardial function depends on the residual myocardial tissue. Thus, response to treatment of acute and chronic MC depends on the specific causes of the disease, severity of irreversible tissue damage at the onset of treatment and the potential of the myocardium to compensate such processes (Figure 2). If pre-treatment damage is severe, aetiology specific treatment options is strongly recommended to prevent rapid progression of the disease but would not achieve significant improvement in ventricular function [196]. Based on current guidelines, the recommended treatment options includes standard HF therapy, antiviral therapy, immunomodulatory therapy and immunosuppression therapy based on the absence of biopsy-proven inflammation, positive PCR viral genomes, and the persistence of HF symptoms > 3 months or > 3 weeks of therapy (Figure 2). For patients with non-responsive HF symptoms, device therapy (implantable cardioverter defibrillator, cardiac resynchronization therapy or ventricular assist device) depending on individual patient characteristics should be considered.
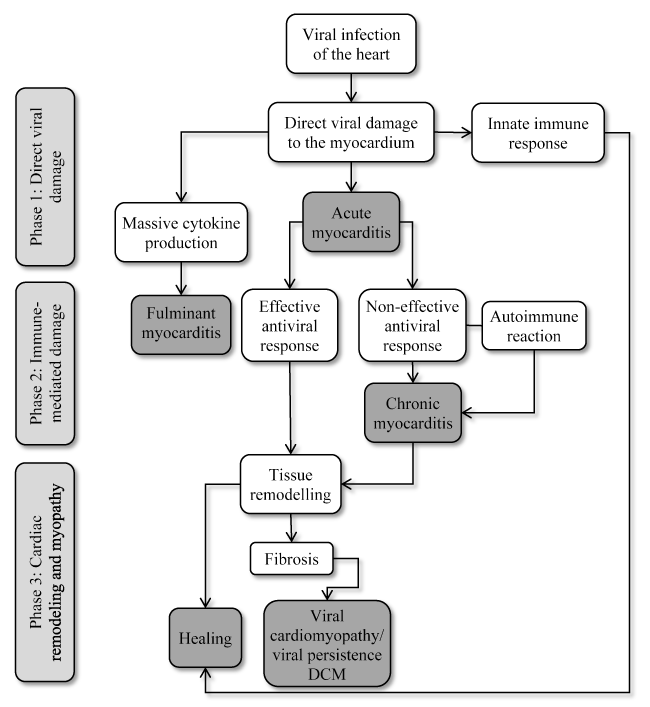
Figure 1. Pathogenesis of Viral Cardiomyopathy
Viral infection of the cardiomyocyte result in direct cytopathic effect leading to cell damage or death. Innate immune response eliminates viral particles and infected cells during the initial virus entry and replication (acute myocarditis). Fulminant myocarditis is rare resulting from disturbances in control mechanism of inflammation. In immunosuppressed patients, ineffective or delayed innate immune response, weak cytotoxic T-lymphocyte response or insufficient antibody production allows viral diffusion within the heart. Chronic viral presence result in constant but still ineffective infiltration of immune cells leading to chronic inflammation. This process accompanied by loss of damaged contractile tissue and the appearance of fibrosis may result in viral cardiomyopathy – a form of dilated cardiomyopathy with persistence viral presence. Modified from Kuffner et al. 2016, p. 395 [60]
Heart failure therapy
The mainstay of treatment for viral MC presenting as DCM with LV systolic dysfunction is the conventional HF medical therapy. The current AHA/ACCF and ESC HF guidelines recommend angiotensin-converting enzyme-inhibitor (ACE-I)/angiotensin receptor blocker (ARBs) and/or beta-blockers [133,226]. Experimental murine myocarditis models show ACE-I (captopril) and ARB (candesartan) relieve HF symptoms in MC patients [227,228]. In patients presenting with myopericarditis-like syndrome of chest pain and normal or near normal of ventricular function, non-steroidal anti-inflammatory drugs such as indomethacin should be avoided because of the risk of increased inflammation and mortality [229]. In addition to guideline-based medical therapy, patients with acute MC should refrain from competitive athletics for 3 to 6 months after diagnosis of viral infection or after documented ventricular recovery by non-invasive imaging modalities [3,196].
Antiarrhythmic therapy
Ventricular arrhythmias are common in patients with active viral heart infection but in most cases do not require specific therapy. However, patients presenting with severe refractory ventricular arrhythmias may require antiarrhythmic therapy while patients with spontaneous remissions may be candidates for long-term antiarrhythmic therapy (amiodarone or ICD) only after all options for controlling arrhythmias have proved unsuccessful [133,226]. Patients with atrioventricular block may need a temporary pacemaker but usually atrioventricular block is transient and indication of permanent pacemaker is rare [226].
Antiviral therapy
The treatment target of antiviral therapy is the elimination of viral translation, transcription and proliferation usually during the initial acute phase of viral infection and replication. Antiviral medication work by preventing viral attachment to host-cells receptors, virus entry or virus uncoating. Medications such as Pleconaril, WIN 54954, or soluble CAR-Fc are only effective during the early stages of the disease. Since, most adults presenting to physicians are in the chronic phase of the disease, the use of antiviral therapy is limited in patients with viral heart diseases. A second challenge of antiviral therapy in patients with viral heart infections is the timing of treatment to prevent progressive myocardial damage by viral clearance before chronic infection causes irreversible damage to myocardial tissues [196,230].
Immunotherapies
Immunotherapies work by modifying immune system to reduce autoimmune-mediated damage to the myocardium during the chronic phase of viral heart infection. Although the evidence to support their use in clinical practice is inconclusive, immunomodulation, immunosuppression and immunoadsorption show promising value in LV function improvement and HF symptoms resolution.
Immunomodulation
Interferon-beta (IFN beta) is an immunomodulatory agent that serve as a natural defence against many viral infections. Innate production of interferon is associated with clinical recovery from viral infection and subsequent sequelae while exogenous administration with protective effect. IFN beta-1 constitute a promising option for treatment for chronic viral heart disease. At present, treatment for chronic viral heart disease is lacking but evidence from uncontrolled open label phase II trials demonstrate sub-groups of patients non-responsive to optimal medical therapy for HF may significantly benefit from 6-months IFN beta-1 medication and HF medication even years after the onset of chronic disease [152,186]. Patients with persistence enterovirus and adenovirus myocardial infection responded well to a 6-months treatment with IFN beta-1 and biopsy-proven complete viral genome elimination three months after termination of antiviral therapy. Viral clearance was accompanied by improved LV function, decreased ventricular size, relief of HF symptoms and decreased infiltrating inflammatory cells [152]. The IFN beta-1 therapy was well tolerated without any unexpected cardiologic or non-cardiologic side effects.
Frequent side effects of IFN beta include influenza-like symptoms and injection site erythema but vanish during the first week of treatment. Patients with severe LV dysfunction (LVEF <25% on IFN beta therapy require echocardiography monitoring because of complaints of mild worsening of HF symptoms due to wall oedema, a slight increase in LV dimension and minor deterioration of LVEF. These complaints disappear within 1 to 2 weeks followed by continuous improvement in HF in about less than 40% of patients [186]. PVB19 and HHV-6 do not affect myocardial contractile cells and thus respond less well on IFN beta treatment with respected to viral clearance and haemodynamic changes. Complete viral clearance for enterovirus and adenovirus suggest that early biopsy-based diagnosis and timely IFN beta treatment may prevent disease progression and consequently outcomes of chronic viral CM [196].
Immunosuppression
Inflammatory processes in the myocardium caused by pathogenic autoimmunity may survive virus elimination in the myocardium and warrant immunosuppressive therapy as a prophylactic for later immune-mediated myocardial damage [225,231,232]. Immunosuppressive therapy requires the exclusion of the virus from the patient based on biopsy findings. Virus-positive patients do not benefit from anti-inflammatory therapy while virus negative patients with post-infectious or autoimmune inflammatory processes respond well. Frequently used anti-inflammatory drugs include immunoglobulins, corticosteroids, azathioprine, and cyclosporine administered for 3 to six months in addition to regular HF medication [225,232]. A randomized trial of 41 patients with immunohistologically biopsy-proven inflammatory CM and 2-year follow-up treated with corticosteroids and azathioprine for three months show sustained benefits in HF symptoms, LV dimensional and LVEF [232]. Noutsias et al. trial validated the diagnostic sensitivity and accuracy of the abundance of cell adhesion molecule (CAM) for inflammatory DCM even in the absence of lymphocytic infiltration because of a close functional association between the induction of CAM, and immunocompetent infiltration and cytokine induction [233]. Thus, CAM is a promising criterion for selecting patients who are likely to benefit from immunosuppressive therapy [233]. Current evidence suggest that immunosuppressive therapy in patients with biopsy-proven virus negative inflammatory CM, in addition to standard HF therapy, is both safe and efficacious. However, there is a need for larger trials powered to detect a difference in clinical endpoints such as HF hospitalization, transplantation and mortality.
Immunoadsorption
Current evidence supporting the use of immunoadsorption therapy such as intravenous immunoglobulin (IVIG) in patients with VMC is inconsistent. An earlier study on acute MC patients treated with IVIG associated high doses of IVIG with improved recovery of LV function and a tendency towards better survival during the first year of presentation [234]. However, a recent study comparing IVIG with cortisone (steroid) therapy reports IVIG is ineffective in children [235]. IVIG and cortisone have comparable effect on freedom from death at the first years and at fifth year and on the median time to recovery of LV function, suggesting IVIG alone confers no advantage to steroid therapy alone [235]. Despite inconsistent evidence, the rationale for serial treatment or high doses of immunoadsorption therapy is to lower cardiotoxic antibodies in the patient’s plasma and prevent autoantibodies cross-reactivity with the host’s intracellular antigen (cardiac myosin) that results in damage to myocardial tissue [174]. Favourable hemodynamic outcomes of immunoadsorption in DCM patients have been related to the elimination of functionally active cardiac autoantibodies evident in biopsy-proven reduction of lymphocytic infiltration and CAM expression [236]. Additional studies are warranted to clarify the therapeutic value of immunoadsorption in patients with VMC.
Meta-analysis of diagnosis and management
The importance of diagnostic methods in informing the selection of the most appropriate treatment emerges clearly in the treatment of VCM patients. At present, consensus guidelines by leading cardiology societies (AHA/ACCF/ESC) discourages the use of EMB except in centres with the requisite expertise and experience because of the associated high risks of complications. Instead, they recommend that diagnosis should include a combination of clinical signs and symptoms, and the presence of ECG abnormalities, markers of myocardial necrosis, echocardiographic or MRI markers of cardiac functional/structural abnormalities and/or tissue characterization by MRI. However, the limited utility of EMB undermines effective clinical management strategies since it is the only reliable method for detecting and quantifying viral presence in the myocardium and for providing information useful for the selection of the most appropriate therapy. While standard HF therapy has proved beneficial in VCM patients with cardiac dysfunction, in a subset of VCM patients who have no demonstrable evidence of cardiac dysfunction or who have refractory symptoms despite optimal medical therapy for heart failure, safe and efficacious treatment has remained a major challenge [3,233]. Antiviral drugs, which target viral elimination in the acute phase, are inappropriate since most patients at presentation are already in the chronic stage of the disease [196].
Findings from recent clinical trials suggest immunosuppression including immunomodulation and immunosuppression are a promising complementary treatment to the standard HF therapy with the potential of improving both cardiac function and functional capacity in VCM patients. However, the evidence is insufficient and inconsistent. Three previous systematic review and meta-analyses did not find evidence that immunosuppressive therapy improved survival or cardiac function compared to conventional HF medication or placebo [237-239]. However, in a sub-population of VMC patients, those with virus-negative myocardium or those with persistent HF symptoms greater than six months despite optimal medical therapy for HF might benefit from immunosuppression [239,240]. Data on the value of immunoadsorption in lowering cardiotoxic antibodies (lymphocytic infiltrates) in VCM patients have not had sufficient evidence to support its use in clinical practice [196]. This systematic review and meta-analysis evaluate diagnostic methods (inclusion criteria) used in previous studies for patient selection and treatment effect of immunotherapies on cardiac function, symptoms, functionality and survival. The search strategy, study selection, data extraction and analyses were performed according to the PRISMA guidelines for systematic review and meta-analysis [241]. Twenty-two (22) studies evaluating the diagnosis and treatment of patients with viral CM were included in this meta-analysis (Table 3).
Table 3. Summary of Studies Included in the Meta-analysis
1st Author |
Year |
Study Methodology |
Patients |
Drugs |
Patients/Entry Criteria |
Main Findings |
Parrillo [242] |
1989 |
Randomized controlled |
59 |
Prednisone |
DCM/CHF: Biopsy evidence for inflammation and immoglobulin elevated erythrocyte sedimentation |
Has marginal effect and should not be administered as a standard therapy |
Latham [243] |
1989 |
Randomized controlled |
52 |
Prednisone |
Idiopathic DCM: Symptom duration < 2 years; LVEF ≤ 45% |
No difference in Mortality at 24 months |
Chan [244] |
1991 |
Retrospective |
13 |
Prednisone + Azathioprine |
Paediatric patients with biopsy-proven MC |
Appears useful in improving clinical course and cardiac function with no adverse side effect |
Drucker [245] |
1994 |
Randomized to IVIG or no IVIG |
46 |
Intravenous immunoglobulin |
Acute CHF < 3 months, LVEF dysfunction due to myocarditis |
High dose IVIG in acute MC improved LV function with a tendency of better survival |
Balaji [246] |
1994 |
Retrospective |
10 |
Corticosteroids |
Children with ventricular ectopic rhythm |
Steroid treatment seems to benefit a subset of children with ventricular ectopic rhythms |
Camargo [247] |
1995 |
Randomized controlled |
43 |
Prednisone + Azathioprine/cyclosporine |
Children with DCM and active MC and severe LV dysfunction |
Improves prognosis of children with MC and severe LV dysfunction |
Mason [248] |
1995 |
Randomized controlled |
111 |
Prednisone + Azathioprine or cyclosporine |
Myocarditis and a LVEF < 45% |
No difference in mortality at one year and LVEF changes |
Lee [249] |
1999 |
Retrospective |
36 |
Intravenous corticosteroids |
Paediatric population with histologically confirmed lymphocytic myocarditis |
Improved survival and recovery of LV function |
Ahdoot [250] |
2000 |
Retrospective |
5 |
Monoclonal OKT3 + intravenous immunoglobulin |
Paediatrics with acute MC and LVEF 5% to 20% |
Reverses/inhibits immune response and improves myocardial function |
Gullestad [251] |
2001 |
Randomized to IVIG of placebo |
40 |
Intravenous immunoglobulin |
Chronic HF > 6 months, LVEF < 40% |
Reduced inflammatory effect and improved LV function |
McNamara [252] |
2001 |
Prospective placebo controlled |
62 |
Intravenous immunoglobulin |
DCM, LVEF ≤ 40%, symptoms < 6 months |
In recent onset DCM, IVIG does not improve LVEF |
Staudt [253] |
2001 |
Randomized crossover IA to IG |
25 |
Immunoadsorption/ immunoglobulin |
DCM patients, LVEF < 30%, evidence of autoantibody and inflammation |
IA followed by IG mitigate myocardial inflammation in DCM patients |
Wojnicz [232] |
2001 |
Randomized to immunosuppression or placebo |
84 |
Prednisone + Azathioprine |
Inflammatory DCM + HLA upregulation on biopsy, symptoms > 6 months |
Long-term improvement LV function, NYHA class |
Frustaci [254] |
2003 |
Retrospective responders vs. non-responders |
41 |
Prednisone + Azathioprine |
Biopsy proven virus negative Lymphocytic MC |
Improved LV function and viral clearance |
Kuhl [62] |
2003 |
Retrospective |
22 |
Interferon beta |
LV dysfunction and EMB-evidence of myocardial virus persistence |
Improved LV function and resulted in viral elimination |
English [255] |
2004 |
Retrospective |
41 |
Intravenous immunoglobulin + steroids |
Children with biopsy proven MC and cardiac dysfunction and cardiotropic viral infection |
IVIG alone does not confer advantage to steroid therapy alone |
Wojnicz [256] |
2006 |
Randomized controlled |
74 |
Artovastin |
HF secondary to inflammatory DCM |
Improved LV function |
Frustaci [225] |
2009 |
Randomized placebo controlled |
85 |
Prednisone + Azathioprine |
Virus-negative inflammatory CM |
Improvement in LV function and dimensions |
Klugman [257] |
2010 |
Randomized to IVIG or no IVIG |
216 |
Intravenous immunoglobulin |
Paediatric myocarditis from paediatric discharges |
IVIG did not confer a survival advantage regardless of patient's severity of illness |
Schmidt-Lucke [186] |
2010 |
Randomized to IFN beta and control |
23 |
Interferon beta |
Presence of virus genome and increased endothelial activation, HF symptoms > 6 months |
Reduced endothelial damage |
Escher [258] |
2016 |
Retrospective |
114/53 |
Prednisone + Azathioprine |
EMB-proven virus -ve chronic MC of inflammatory DCM, LVEF< 45% |
Short/long term improvement in LVEF and reduction in inflammatory infiltrates |
Merken [259] |
2018 |
Randomized controlled |
180 |
Prednisone + Azathioprine |
Patients With virus-negative non-fulminant CM, symptoms < 6 months |
Improved heart transplant-free survival and larger improvement in LVEF |
CM: Cardiomyopathy; DCM: Dilated Cardiomyopathy; EMB: Endomyocardial Biopsy; HF: Heart Failure; IA: Immunoadsorption; IFN: Interferon; IVIG: Intravenous Immunoglobulin; LV: Left Ventricular; LVEF: Left Ventricular Ejection Fraction; MC: Myocarditis
Findings
In total, the 22 studies included in this meta-analysis enrolled 1,321 patients with either VMC or VCM with mean age of 33 years (range 3 to 60 years) with a greater proportion of male patients (60%) [62, 186,225,232,242-259]. Fourteen (14) studies were randomized controlled trialswhile the remaining eight (8) were retrospective cohort studies [62,225,232,242-244,246-249,250-259]. While randomized control trials provide strong evidence, observational studies provide actual experience of the application of findings in a clinical setting. Twelve studies assessed immunosuppression (prednisone, azathioprine, cyclosporine or corticosteroids), seven assessed immunoadsorption (immunoglobulin) and the remaining studies assessed immunomodulation (interferon beta) [62,186, 225,232,242-249,252-259]. Outcomes measured varied across studies with LVEF being a common feature followed by LV dimensions. Other common measures assessed in more than two studies included NYHA functional class, viral genomes clearance, and death and hospitalization.
Various diagnostic or inclusion criteria for patient selection were used by individual studies. However, the most common features included LV systolic dysfunction (measured as LVEF), the duration of symptoms usually longer than six months, refractory symptoms and biopsy-proven virus-free myocardium. In more than two studies, common clinical presentation were fever, shortness of breath, chest pain, palpitations (arrhythmias), pre-syncope or syncope (atrioventricular block), arrhythmia and HF. In almost all the studies, EMB was the most common method for quantifying inflammatory infiltrates (viral genomes), and together with symptoms duration (>6 months) was used as part of selection criterion in four studies [225,232,254,258]. Prolonged symptom duration potentially indicates non-responsiveness to HF therapy. However, cut off values of LV systolic dysfunction (LVEF) varied from as low as ≤ 30% to as high as ≤ 45%, and one study included DCM patients with HLA upregulation on biopsy specimens [[135,150,232].
Pooled analysis of 11 studies showed that the addition of immunosuppression therapies to standard HF therapy significantly improved LV systolic function in the short-term (6-12 months) [62,225,232,242,246,251,253,254,256,258,259]. Weighted mean difference (WMD) of LVEF at baseline and after 6 or 12 months of immunosuppressive therapy revealed a significant increase in LVEF (WMD: 8.61% 95% CI: 5.54% to 11.66%; p = 0.000). (Figure 3). A greater improvement in LV function was also observed in patients receiving immunosuppressive therapy compared to patients receiving only standard HF therapy or placebo (WMD: 9.97%; 95% CI: 3.78% to 16.16%) (Figure 4). Immunosuppression was also associated with significant improvement in LV dimensions observed as reductions in LV end diastolic diameter (LVEDD) and volume (LVEDV) at baseline relative to after six months of treatment. LVEDD reduced by a weighted mean of -5.57 mm (95% CI: -8.85 to -2.29; p = 0.001) (Figure 5) and LVEDV reduced by -56.60 ml (95% CI: -88.82 to -24.39; p = 0.001) (Figure 6). However, there was significant heterogeneity across individual studies due to differences in the characteristics of enrolled patients, the extent of depressed LVEF, the duration of symptoms, age of patients, myocarditis or DCM patients and the presence or the absence of inflammatory infiltrates in the myocardium. The effect of these differences were not considered in the present meta-analysis.
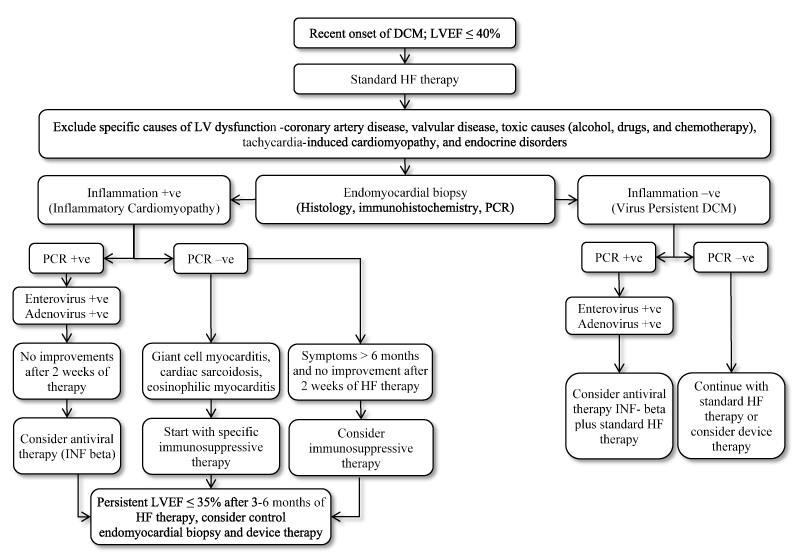
Figure 2. Diagnostic and Therapeutic Algorithm for Suspected Inflammatory DCM Adapted from Krejci et al., 2016 [58]
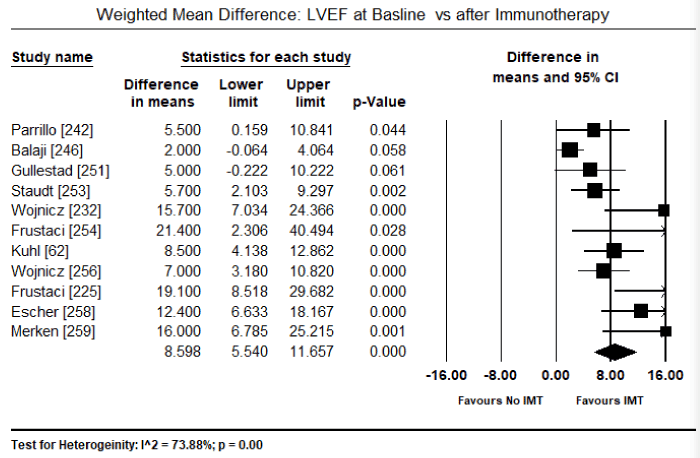
Figure 3. Forest Plot of LVEF Pre and Post-Immunotherapy
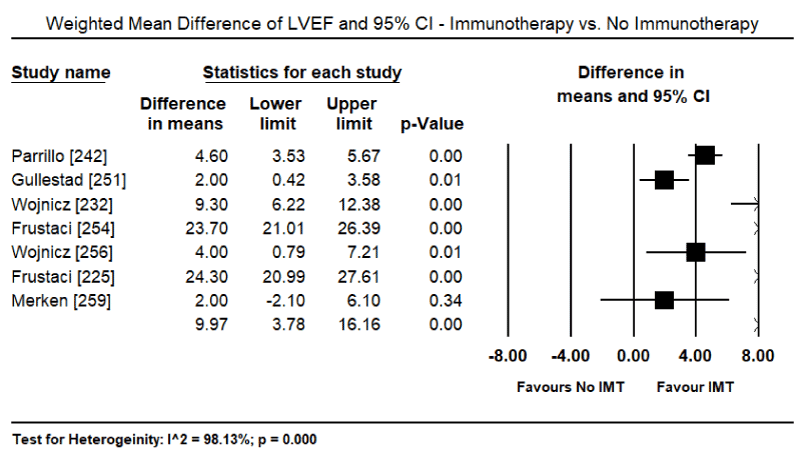
Figure 4. Forest Plot for LVEF Differences - Immunotherapy and No Immunotherapy
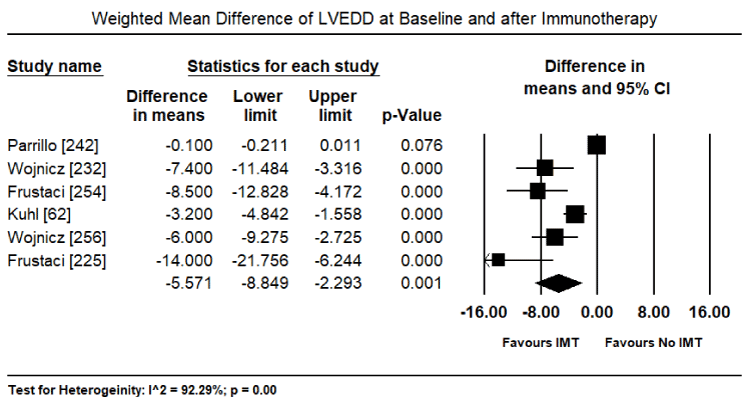
Figure 5. Forest Plot of LVEDD Pre and Post-Immunotherapy
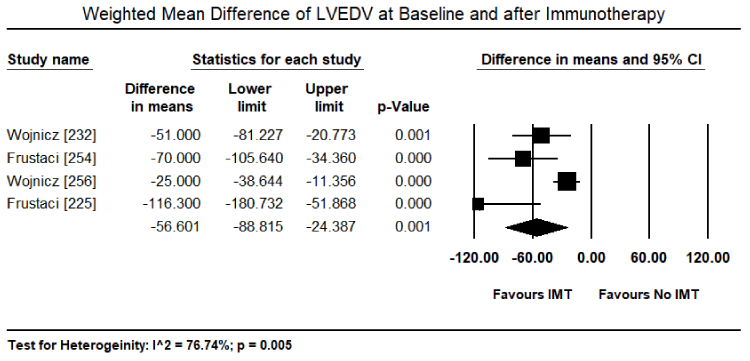
Figure 6. Forest Plot of LVEDV Pre and Post- Immunotherapy
Pooled analysis of four studies reveal a tendency of immunosuppressive therapy improvement in cardiac function compared to patients on conventional therapy [232,242,251,256]. There was a reduction in the weighted mean of NYHA functional class (WMD: -1.76; 95% CI: -3.74 to 0.21) but it was non-significant (p = 0.08) (Figure 7). The significant heterogeneity across individual studies could be attributed to the inclusion of patients from all the functional groups (NYHA I – IV), since NYHA it was not part of selectin criterion in most of the included studies. Pooled analysis of three studies show immunosuppression therapy significantly reduced inflammatory infiltrates in the myocardium six months after commencing therapy (WMD: -5.90; 95% CI: -10.90 to -0.89) (Figure 8) [62,253,258]. The effect of immunosuppression on hard clinical endpoint of death and hospitalization or heart transplantation show a tendency towards a reduction in the events. Pooled analysis of six studies showed a non-significant reduction by 10% of deaths in the short-term (6 to 12 months) compared to conventional therapy or placebo (odds ratio [OR]: 0.90; 95% CI: 0.57 to 1.43; p = 0.67) (Figure 9) [225,248,252,255,257,259]. Pooled analysis of four studies also reveal a non-significant reduction of hospitalization by 20% in the short-term (6 to 12 months) compared to conventional HF therapy or placebo (OR: 0.80; 95% CI: 0.47 to 1.38; p = 0.43) (Figure 10) [225,252,255,259]. There was no heterogeneity observed across studies while analyzing deaths and hospitalization.
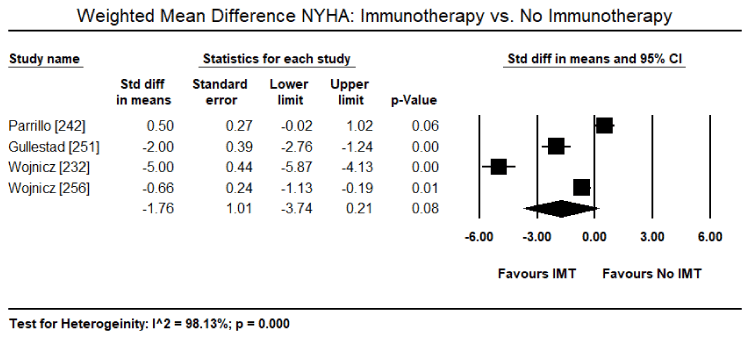
Figure 7. Forest Plot for NYHA Differences - Immunotherapy and No Immunotherapy
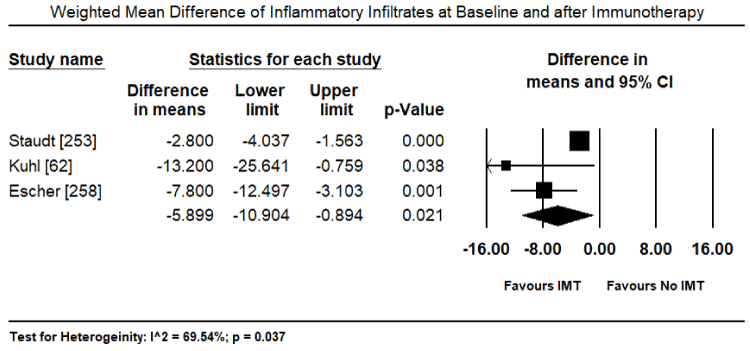
Figure 8. Forest Plot of Inflammatory Infiltrates Pre and Post-Immunotherapy
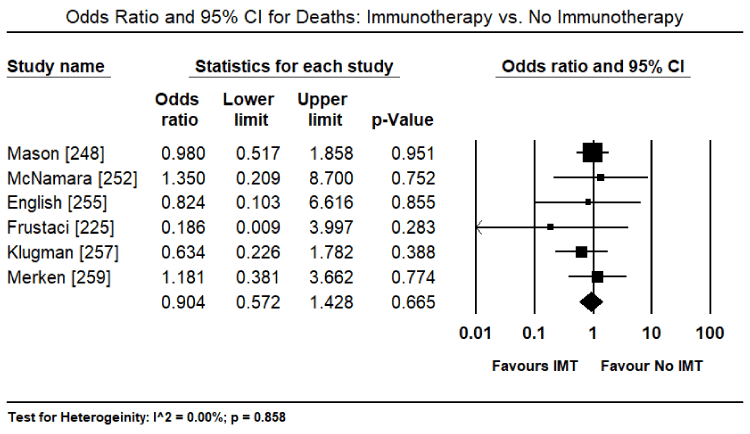
Figure 9. Forest Plot for Deaths – Immunotherapy vs. No Immunotherapy
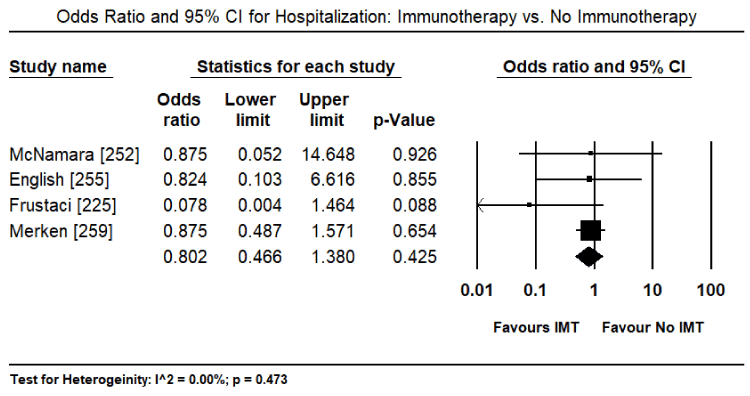
Figure 10. Forest Plot for Hospitalization – Immunotherapy vs. No Immunotherapy
Discussion
Clinical Investigations
Viral infection is the most common aetiology of infective MC, which remains a complex and challenging diagnosis in cardiology. Recent consensus guidelines recommend diagnosis should be based on clinical presentation and the presence of ECG abnormalities accompanied by biomarkers of myocardial inflammation and/or non-invasive evidence of abnormalities in cardiac structure and/or function [3,205]. Although in the present meta-analysis clinical signs and symptoms of viral CM could not be pooled because insufficient numerical data was provided in the individual studies, common clinical presentation mentioned in more than two studies were fever, shortness of breath, chest pain, palpitations (arrhythmias), pre-syncope or syncope (atrioventricular block), arrhythmia and HF. These signs and symptoms of VCM are consistent with those reported in previous studies although HF, chest pain and arrhythmias have been cited as the most common reason for hospital visitation [205-207]. Most of the enrolled patients in the individual studies had persistence symptoms for more than six months suggesting worsening cardiac function and the likelihood to benefit from immunosuppression. These symptoms were typical of cardiac dysfunction while symptoms of the underlying viral disease or infection were not described in the individual studies. In addition to clinical presentation, common ECG abnormalities in the included studies were T-wave inversion, ST-segment elevation, ST-segment depression, PR depression, and pathologic Q waves. However, ECG abnormalities have been described previously as non-specific to VCM patients since they may mimic other cardiac disorders such as acute myocardial infarction or pericarditis [260]. Clinical presentation and ECG abnormalities are useful for raising clinical suspicion for viral MC and CM, while EMB is the most common test for confirming diagnosis as well as patient selection in almost all the included studies.
Consistent with literature on MC, standardized histopathological description of biopsy sample was used to detect myocardial inflammation associated with necrosis in the absence of ischemia in MC patients [260]. However, histopathology alone has been found to be inadequate to detect the presence of active MC. It is too narrow due to limited variable interpretation, lacks clinical prognostic value, and has poor diagnostic sensitivity and specificity [261]. To improve histological diagnosis of viral MC, additional virological and immunological evaluation of myocardial tissues have been recommended using immunohistochemical and PCR techniques, which allow the identification and quantification of inflammatory cells and viral infection markers [261]. Although EMB has been the gold standard for confirmatory diagnosis of infective MC and DCM for long, evidence is emerging that cardiac MRI is an important tool for non-invasive evaluation of patients suspected with MC [220,262,263]. In the diagnosis of VCM, cardiac MRI is used to evaluate functional and morphological abnormalities as well as tissue pathology as a feature of myocardial inflammation. With a lack of consensus on EMB, a recent AHA consensus statement recommends that EMB should be reserved for patients who are likely to have specific myocardial disorders with unique prognoses and specific treatment recommendations [264]
Clinical management
In the present systematic review and meta-analysis, pooled analysis of five studies (N = 406) reveal statistical evidence of beneficial uses of immunosuppressive therapy in addition to standard HF therapy in improving LV systolic function and cardiac hemodynamics (reduction of LV volumes and dimensions). Immunosuppressive therapy also revealed a trend towards reduction in death, and HF hospitalization and/or heart transplantation. Since these benefits are only likely to occur in a sub-group of VMC patients, it underscores the importance of diagnostic methods in the selection of patients who will benefit from treatment. These patients include those with biopsy-proven virus free myocardium in the presence or absence of myocardial inflammation, echocardiographic evidence of depressed systolic function (LVEF ≤45%), and refractory HF symptoms greater than six months despite optimal medical therapy for HF. The importance of patient selection emerged from previous four meta-analyses in which the included studies enrolled patients without evidence of biopsy-proven virus negative myocardium and with HF symptoms less than six months [144-147]. In this meta-analysis, studies reported no evidence of immunosuppressive therapy improving hemodynamics and cardiac function relative to conventional HF therapy or placebo.
The evidence of therapeutic benefits of immunosuppressive therapy is consistent with findings of animal models and proposed tri-phasic pathogenic mechanisms of VMC. The model proposes that VMC begins with an acute phase of viral infection and replication leading to direct viral damage to the myocardium followed by the phase of immune-mediated response to viral infection and its damaging effect on the myocardium due to molecular mimicry and the final phase of cardiac remodelling [55-61]. Immunosuppressive therapy targets the second phase of immune-mediated myocardial damage by suppressing immune response. Patients with biopsy-proven virus free myocardium benefit from immunosuppression because after the completion of the virus-killing phase, the action of autoantibodies on the cardiomyocytes may persist through molecular mimicry in which immunosuppressive drugs reduce cardiomyocyte destruction and prevent its progression to VMC [152,153]. Thus, timing of immunosuppressive therapy is critical because its administration during viral infection and replication phase would suppress the host immune response with deleterious effect on cardiac function. While some studies suggest a positive trend towards immunomodulation and immunoadsorption improving cardiac function in VCM patients in the present analysis, there was no sufficient data to perform a pooled analysis and determine therapeutic value of VCM [152,186,234,235],
Limitations
Despite the present findings supporting the use of immunosuppressive therapy in a sub-group of VMC patients, a few limitations in this meta-analysis should be considered. Most of the data on the use of immunosuppressive therapy have been gathered from animal models or autopsy analysis and extrapolated to the general population while individual large-scale randomized clinical trials are difficult to conduct. This has resulted in the current small sample sizes of the included studies, and heterogeneity in terms of diagnosis, methodology, treatment protocols and outcomes measures. Studies with larger samples are needed to enhance the statistical power detect significant differences in treatment effect for individual studies. The criteria for patient selection into trials is not clearly defined. Most of the patients selected had cardiac symptoms >6 months and biopsy-proven virus free myocardium and thus the findings cannot be applied to all patients with viral infection of the myocardium. Viral MC of DCM is a disease difficult to diagnose at initial presentation because its clinical presentation is protean and with a wide spectrum ranging from asymptomatic phase to acute cardiovascular collapse requiring mechanical support. Moreover, clinical and histological features may overlap with that of inflammatory cardiomyopathy [3]. Upregulated HLA expression in the myocardium is a promising criterion to identify patients who would benefit from immunosuppressive therapy but used by only one study [142].
Clinical implications
The current findings have important clinical implications. Although the findings support the use of immunosuppression, it is based on small-scale individual studies with short-term outcomes that has prevented broad application of its benefits to clinical settings. Small-scale trials might lack the statistical power to detect significant differences in important clinical end-points such as recurrent heart failure, the need for ventricular assist devices, heart transplantation or death. While larger trials with carefully selected patients are needed to confirm the benefits of immunosuppression, its use in a larger population has the potential to cause a measureable increase in immunosuppression-related side effects, infections and possibly malignancies [265]. Larger trials should also have longer follow-up periods to enable prospective evaluations since short-term benefits such as in LV systolic function (LVEF) may not correlate with long-term risk of death or heart transplantation in the subset of VCM patients who are likely to benefit from immunosuppression. Although the use of cellular HLA antigen or adhesion molecule are promising to identify VMC patients who might benefit from immunosuppression, there is no sufficient evidence to support their use in clinical setting. Despite the clinical benefits of immunosuppression based on biopsy evidence of virus negative myocardium, its use is limited because on a small minority of medical centres that possess the capability to perform endomyocardial biopsy and the expertise in cardiac pathology and viral genome analysis. Finally, the present findings would stimulate research for the effect of other medications targeting the immune mechanisms such as immunoadsorption in the prevention of immune-mediate myocardial damage in VMC patients during the chronic phase of the disease.
Conclusion
Viral cardiomyopathy (VCM) is a myocardial disease due to viral persistence in a dilated heart in the absence of myocardial inflammation. Its exact epidemiology is unknown due to wide heterogeneity of causative viruses and periods of epidemic that alter its prevalence and incidence. The main pathogenic mechanisms are viral and immune-mediated damages to the myocardium leading to myocardial remodelling, or damage to the vascular endothelial cells leading to microvascular dysfunction or the degradation of the interstitial matrix causing LV dilatation and dysfunction. Typical manifestation include chest pains, arrhythmia or heart failure. Endomyocardial biopsy remains the diagnostic gold standard but increased risk of complications and the need for expert knowledge limits its widespread use. The criteria for clinical diagnosis is a combination of symptoms, ECG abnormalities and non-invasive imaging evidence of structural/functional dysfunction. The available treatment strategies include HF therapy for the relief of symptoms and antiarrhythmic therapy for the management of ventricular arrhythmias. In addition to HF medication, immunosuppressive therapy in biopsy-proven virus free myocardium with refractory HF symptoms improves cardiac function and hemodynamics. However, the use of immunosuppressive is limited by insufficient supporting evidence and the lack of expertise needed for heart biopsy. Large-scale studies with a long follow-up period and enrolling carefully selected patients would help to determine long-term outcomes of hard clinical endpoints such as death, hospitalization and heart transplantation to encourage broad adoption of immunosuppression in clinical practice.
References
- Richadson P (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology. Task force on the definition and classification of cardiomyopathies. Circulation 93: 841-842. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F et al. (2008) Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur Heart J 29: 270-276. [Crossref]
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. 113: 1807-1816. [Crossref]
- Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B et al. (2013) The MOGE (S) classification for a phenotype–genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol 62: 2046-2072. [Crossref]
- Arbustini E, Narula N, Tavazzi L, Serio A, Grasso M et al. (2014) The MOGE (S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol 64: 304-318. [Crossref]
- Maisch B, Ristic AD, Portig I, Pankuweit S (2003) Human viral cardiomyopathy. Front Biosci 8: s39-s67. [Crossref]
- Schichao LV, Rong J, Ren S, Wu M, Li M et al. (2013) Epidemiology and diagnosis of viral myocarditis. Hellenic J Cardiol 54: 382-391. [Crossref]
- Wakafuji S, Okada R (1986) Twenty Year Autopsy Statistics of Myocarditis Incidence in Japan: the 10th Conference on prevention for rheumatic fever and rheumatic heart disease. Jpn Circ J 50: 1288-1293. [Crossref]
- Passarino G, Burlo P, Ciccone G, Comino A (1997) Prevalence of myocarditis at autopsy in Turin, Italy. Arch Pathol Lab Med 121: 619-622. [Crossref]
- Hufnagel G, Pankuweit S, Richter A, Schönian U, Maisch B et al, (2000) The European Study of Epidemiology and Treatment of Cardiac Inflammatory Diseases (ESETCID) First Epidemiological Results. Herz 25: 279-285. [Crossref]
- Ukimura A, Satomi H, Ooi Y, Kanzaki Y (2012) Myocarditis Associated with Influenza A H1N1pdm2009. Influenza Res Treat 2012:351979 [Crossref]
- Sotiriou E, Heiner S, Jansen T, Brandt M, Schmidt KH et al. (2018) Therapeutic implications of a combined diagnostic workup including endomyocardial biopsy in an all‐comer population of patients with heart failure: a retrospective analysis. ESC Heart Fail 5: 630-641[Crossref]
- Kyto V, Saraste A, Voipio-Pulkki LM, Saukko P (2007) Incidence of fatal myocarditis: a population-based study in Finland. Am J Epidemiol 165: 570-574. [Crossref]
- Lv S, Rong J, Ren S, Wu M, Li M et al. (2013) Epidemiology and diagnosis of viral myocarditis. Hellenic J Cardiol 54: 382-391. [Crossref]
- Marboe CC, Fenoglio Jr JJ (1988) Pathology and natural history of human myocarditis. Pathol Immunopathol Res 7: 226-239. [Crossref]
- Le Gall O, Christian P, Fauquet CM, King AM, Knowles NJ et al. (2008). Picornavirales, a proposed order of positive-sense single-stranded RNA viruses with a pseudo-T= 3 virion architecture. Arch Viro. 153: 715-727. [Crossref]
- Cabrerizo M, Tarrago D, Muñoz‐Almagro C, Del Amo E, Domínguez‐Gil M et al. (2014) epidemiology of enterovirus 71, coxsackievirus A 16 and A 6 associated with hand, foot and mouth disease in Spain. Clin Microbiol Infect 20: O150-O156. [Crossref]
- Osterback R, Vuorinen T, Linna M, Susi P, Hyypiä T et al. (2009) Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis 15: 1485-1488. [Crossref]
- Fujimoto T, Iizuka S, Enomoto M, Abe K, Yamashita K et al. (2012) Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis 18: 337-339. [Crossref]
- Modlin JF, Rotbart HA (1997) Group B coxsackie disease in children. Curr Top Microbiol Immunol 223: 53-80. [Crossref]
- Palacios G, Oberste MS (2005) Enteroviruses as agents of emerging infectious diseases. J Neurovirol 11: 424-433. [Crossref]
- Remes J, Helin M, Vaino P, Rautio P (1990) Clinical outcome and left ventricular function 23 years after acute Coxsackie virus myopericarditis. Eur Heart J 11: 182-188. [Crossref]
- See DM, Tilles JG (1991) Viral myocarditis. Rev Infect Dis13: 951-956. [Crossref]
- Hingorani AD (1992) Postinfectious myocarditis. BMJ 304: 1676-1678. [Crossref]
- Eggers HJ (1990) Notes on the pathogenesis of enterovirus infections. Observations, experiments, and speculations. Medical microbiology and immunology 179: 297-306. [Crossref]
- Waterson AP (1978) Virological investigations in congestive cardiomyopathy. Postgrad Med 54: 505-507. [Crossref]
- Kitaura Y (1981) Virological study of idiopathic cardiomyopathy: serological study of virus antibodies and immunofluorescent study of myocardial biopsies. Jpn Circ J 45: 279-294. [Crossref]
- Fujioka S, Kitaura Y, Ukimura A, Deguchi H, Kawamura K et al. (2000) Evaluation of viral infection in the myocardium of patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 36: 1920-1926. [Crossref]
- Frustaci A, Maseri A (1992) Localized left ventricular aneurysms with normal global function caused by myocarditis. Am J Cardiol 70: 1221-1224. [Crossref]
- Noutsias M, Fechner H, de Jonge H, Wang X, Dekkers D et al. (2001) Human coxsackie-adenovirus receptor is colocalized with integrins αvβ3 and αvβ5 on the cardiomyocyte sarcolemma and upregulated in dilated cardiomyopathy: implications for cardiotropic viral infections. Circulation 104: 275-280. [Crossref]
- Heegaard ED, Brown KE (2002) Human parvovirus B19. Clin Microbiol Rev 15: 485-505. [Crossref]
- Sharad S, Kapur S (2005) Emerging human infections: An overview on Parvovirus B19. J Indian Acad Clin Med 6: 319-326.
- Pankuweit S, Portig I, Eckhardt H, Crombach M, Hufnagel G et al. (2000) Prevalence of viral genome in endomyocardial biopsies from patients with inflammatory heart muscle disease. Herz 25: 221-226. [Crossref]
- Jiang SC (2006) Human adenoviruses in water: occurrence and health implications: a critical review. Environ Sci Technol 40: 7132-7140. [Crossref]
- Ghebremedhin B (2014) Human adenovirus: Viral pathogen with increasing importance. Eur J Microbiol Immunol 4: 26-33. [Crossref]
- Russell WC (2009) Adenoviruses: update on structure and function. J Gen Virol virology 90: 1-20. [Crossref]
- Lion T (2014) Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev 27: 441-462. [Crossref]
- Davison AJ, Benko M, Harrach B (2003) Genetic content and evolution of adenoviruses. J Gen Virol 84: 2895-2908. [Crossref]
- Pauschinger M, Bowles NE, Fuentes-Garcia FJ, Pham V, Kühl U et al. (1999) Detection of adenoviral genome in the myocardium of adult patients with idiopathic left ventricular dysfunction. Circulation 99: 1348-1354. [Crossref]
- Braun DK, Dominguez G, Pellett PE (1997) Human herpesvirus 6. Clin Microbiol Rev 10: 521-567. [Crossref]
- Agut H, Bonnafous P, Gautheret-Dejean A (2015) Laboratory and clinical aspects of human herpesvirus 6 infections. Clin Microbiol Rev 28: 313-335. [Crossref]
- Yao K, Crawford JR, Komaroff AL, Ablashi DV, Jacobson S (2010) Review part 2: Human herpesvirus-6 in central nervous system diseases. J Med Virol 82: 1669-1678. [Crossref]
- Caserta MT, Mock DJ, Dewhurst S (2001) Human herpesvirus 6. Clin Infect Dis 33: 829-833. [Crossref]
- Kuhl U, Pauschinger M, Noutsias M, Seeberg B, Bock T et al. (2005) High prevalence of viral genomes and multiple viral infections in the myocardium of adults with" idiopathic" left ventricular dysfunction. Circulation 111: 887-893. [Crossref]
- Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M et al. (2005) Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation 112: 1965-1970. [Crossref]
- Yoshikawa T, Ihira M, Suzuki K, Suga S, Kito H et al. (2001) Fatal acute myocarditis in an infant with human herpesvirus 6 infection. J Clin Pathol 54: 792-795. [Crossref]
- Reddy S, Eliassen E, Krueger GR, Das BB (2017) Human herpesvirus 6-induced inflammatory cardiomyopathy in immunocompetent children. Ann Pediatr Cardiol 10: 259-268. [Crossref]
- Stefanski HE, Thibert KA, Pritchett J, Prusty BK, Wagner JE et al. (2016) Fatal myocarditis associated with hhv-6 following immunosuppression in two children. Pediatrics 137. [Crossref]
- Schottstedt V, Blümel J, Burger R, Drosten C, Gröner A et al. (2010) Human cytomegalovirus (HCMV)–revised. Transfus Med Hemother 37: 365-375. [Crossref]
- Ljungman P, Griffiths P, Paya C (2002) Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis 34: 1094-1097. [Crossref]
- Lowry RW, Adam E, Hu C, Kleiman NS, Cocanougher B et al. (1994) What are the implications of cardiac infection with cytomegalovirus before heart transplantation? J. Heart Lung Transplant 13: 122-128. [Crossref]
- Partanen J, Nieminen MS, Krogerus L, Geagea A, Lautenschlager I et al. (1991) Cytomegalovirus myocarditis in transplanted heart verified by endomyocardial biopsy. Clin Cardiol 14: 847-849. [Crossref]
- Gonwa TA, Capehart JE, Pilcher JW, Alivizatos PA (1989) Cytomegalovirus myocarditis as a cause of cardiac dysfunction in a heart transplant recipient. Transplantation 47: 197-199. [Crossref]
- Shabtai M, Luft B, Waltzer WC, Anaise D, Rapaport FT (1988) Massive cytomegalovirus pneumonia and myocarditis in a renal transplant recipient: successful treatment with DHPG. Transplant Proc 20: 562-563. [Crossref]
- Schindler JM, Neftel KA (1990) Simultaneous primary infection with HIV and CMV leading to severe pancytopenia, hepatitis, nephritis, perimyocarditis, myositis, and alopecia totalis. Klin Wochenschr 68: 237-240. [Crossref]
- Arvin AM (1996) Varicella-zoster virus. Clin Microbiol Rev 9: 361-381. [Crossref]
- Gershon AA, Breuer J, Cohen JI, Cohrs RJ, Gershon MD et al. (2015) Varicella zoster virus infection. Nat Rev Dis Primers 1:15016. [Crossref]
- Gershon AA, Gershon MD (2013) Pathogenesis and current approaches to control of varicella-zoster virus infections. Clin Microbiol Rev 26: 728-743. [Crossref]
- Tsintsof A, Delprado WJ, Keogh AM (1993) Varicella zoster myocarditis progressing to cardiomyopathy and cardiac transplantation. Heart 70: 93-95. [Crossref]
- Rich R, McErlean M (1993) Complete heart block in a child with varicella. Am J Emerg Med 11: 602-605. [Crossref]
- Ioannou A, Tsappa I, Metaxa S, Missouris CG (2017) Ventricular Fibrillation following Varicella Zoster Myocarditis. Case Rep Cardiol 2017: 1017686. [Crossref]
- Kim CW, Chang KM (2013) Hepatitis C virus: virology and life cycle. Clin Mol Hepatol 19: 17-25. [Crossref]
- Li HC, Lo SY (2015) Hepatitis C virus: Virology, diagnosis and treatment. World J Hepatol 7: 1377-1389. [Crossref]
- Bukh J (2016) The history of hepatitis C virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J Hepatol 65: S2-S21. [Crossref]
- Matsumori A, Yutani C, Ikeda Y, Kawai S, Sasayama S (2000) Hepatitis C virus from the hearts of patients with myocarditis and cardiomyopathy. Lab Invest 80: 1137-1142. [Crossref]
- Singh DS, Gupta PR, Gupta SS, Bhatia PK, Somani PN et al. (1989) Cardiac changes in acute viral hepatitis in Varanasi (India). J Trop Med Hyg 92: 243-248. [Crossref]
- Ursell PC, Habib A, Sharma P, Mesa-Tejada R, Lefkowitch JH et al. (1984). Hepatitis B virus and myocarditis. Human Pathology 15: 481-484. [Crossref]
- Boyella V, Onyebueke I, Farraj N, Graham-Hill S, El Younis C et al. (2009) Prevalence of hepatitis C virus infection in patients with cardiomyopathy. Annals of hepatology. 8: 113-115. [Crossref]
- Matsumori A (2000) Hepatitis C virus and cardiomyopathy. Herz 25: 249-254. [Crossref]
- Szewczyk B, Bieńkowska-Szewczyk K, Król E (2014) Introduction to molecular biology of influenza a viruses. Acta Biochim Pol 61: 397-401 [Crossref]
- Noda T (2012) Orthomyxoviruses. Uirusu 62: 219-228. [Crossref]
- Blut A (2009) Influenza virus. Transfus Med Hemothe 36: 32-39. [Crossref]
- Sprenger MJ, Van Naelten MA, Mulder PG, Masurel N (1989) Influenza mortality and excess deaths in the elderly, 1967-82. Epidemiol Infect 103: 633-641. [Crossref]
- Herskowitz A, Campbell S, Deckers J, Kasper EK, Boehmer J et al. (1993). Demographic features and prevalence of idiopathic myocarditis in patients undergoing endomyocardial biopsy. Am J Cardiol 71: 982-986. [Crossref]
- Chan KY, Iwahara M, Benson LN, Wilson GJ, Freedom RM (1991) Immunosuppressive therapy in the management of acute myocarditis in children: a clinical trial. J Am Coll Cardiol 17: 458-460. [Crossref]
- Maisch B, Ristic AD, Hufnagel G, Pankuweit S (2002) Pathophysiology of viral myocarditis: the role of humoral immune response. Cardiovasc Pathol 11: 112-122. [Crossref]
- Wimmer E, Hellen CU, Cao X (1993) Genetics of poliovirus. Annu Rev Genet 27: 353-436. [Crossref]
- Clough P (1951) Myocarditis in poliomyelitis and related viral infections. Ann Intern Med 34: 1502-1508. [Crossref]
- Minor PD (2004) Polio eradication, cessation of vaccination and re-emergence of disease. Nat Rev Microbiol 2: 473-482. [Crossref]
- Weinstein L (1957) Cardiovascular disturbances in poliomyelitis. Circulation 15: 735-756. [Crossref]
- Laake H (1951) Myocarditis in poliomyelitis. Acta Med Scand 140: 159-169. [Crossref]
- Spain DM, Bradess VA, Parsonnet V (1950) Myocarditis in poliomyelitis. Am Heart J 40: 336-344. [Crossref]
- Rubin S, Eckhaus M, Rennick LJ, Bamford CG, Duprex WP (2015) Molecular biology, pathogenesis and pathology of mumps virus. J Pathol 235: 242-252. [Crossref]
- Rosenberg D (1945) Acute myocarditis in mumps (epidemic parotitis). Arch Intern Med (Chic) 76: 257-263. [Crossref]
- Hussain S, Zahid MF, Rahman AJ, Ahmed MA, Ibrahim SH (2016) Mumps myocarditis: a forgotten disease? J Ayub Med Coll Abbottabad 28: 201-203. [Crossref]
- Lee JY, Bowden DS (2000) Rubella virus replication and links to teratogenicity. Clin Microbiol Rev 13: 571-587. [Crossref]
- Lambert N, Strebel P, Orenstein W, Icenogle J, Poland GA (2015) Rubella. Lancet 385: 2297-2307. [Crossref]
- Ainger LE, Lawyer NG, Fitch CW (1966) Neonatal rubella myocarditis. Br Heart J 28: 691-697. [Crossref]
- Leaver M (1998) Measles (Rubeola) virus. Collegian 5: 40-41. [Crossref]
- Laksono B, de Vries R, McQuaid S, Duprex W, de Swart R (2016) Measles virus host invasion and pathogenesis. Viruses 8: 210. [Crossref]
- Naim HY (2015) Measles virus: a pathogen, vaccine, and a vector. Hum Vaccin Immunother11: 21-26. [Crossref]
- Frustaci A, Abdulla AK, Caldarulo M, Buffon A (1990) Fatal measles myocarditis. Cardiologia 35: 347-349. [Crossref]
- Finkel HE (1964). Measles myocarditis. Am Heart J 67: 679-683. [Crossref]
- Babkin I, Babkina I (2015) The origin of the variola virus. Viruses 7: 1100-1112. [Crossref]
- Li Y, Carroll DS, Gardner SN, Walsh MC, Vitalis EA et al. (2007) On the origin of smallpox: correlating variola phylogenics with historical smallpox records. Proc Natl Acad Sci 104: 15787-15792. [Crossref]
- Keinath K, Church T, Kurth B, Hulten E (2018) Myocarditis secondary to smallpox vaccination. BMJ Case Rep bcr-2017. [Crossref]
- Cassimatis DC, Atwood JE, Engler RM, Linz PE, Grabenstein JD et al. (2004) Smallpox vaccination and myopericarditis: a clinical review. J Am Coll Cardiol 2004 May 5;43: 1503-1510. [Crossref]
- Stanfield BA, Luftig MA (2017) Recent advances in understanding Epstein-Barr virus. F1000Research. 6: 386. [Crossref]
- Young LS, Rickinson AB (2004) Epstein–Barr virus: 40 years on. Nat Rev Cancer 4: 757-768. [Crossref]
- Cohen JI (2000) Epstein–Barr virus infection. New Eng J Med 343: 481-492. [Crossref]
- Von Olshausen G, Hyafil F, Langwieser N, Laugwitz KL, Schwaiger M et al. (2014) Detection of acute inflammatory myocarditis in Epstein Barr virus infection using hybrid 18F-fluoro-deoxyglucose–positron emission tomography/magnetic resonance imaging. Circulation 130: 925-926. [Crossref]
- Ishikawa T, Zhu BL, Li DR, Zhao D, Maeda H (2005) Epstein–Barr virus myocarditis as a cause of sudden death: two autopsy cases. Int J Legal Med 119: 231-235. [Crossref]
- Fraisse A, Paut O, Zandotti C, Lagier P, Camboulives J et al. (2000) An unusual cause of acute myocarditis in children. Arch Pediatr 7: 752-755. [Crossref]
- López SZ, Vicario JM, Lerín FJ, Fernández A, Pérez G et al. (2010) Epstein-Barr virus myocarditis as the first symptom of infectious mononucleosis. Intern Med 49 :569-571. [Crossref]
- Carrol ED, Beadsworth MB, Jenkins N, Ratcliffe L, Ashton I et al. (2006) Clinical and diagnostic findings of an echovirus meningitis outbreak in the north west of England. Postgrad Med J 82: 60-64. [Crossre]
- Bernit E, de Lamballerie X, Zandotti C, Berger P, Veit V et al. (2004) Prospective investigation of a large outbreak of meningitis due to echovirus 30 during summer 2000 in Marseilles, France. Medicine (Baltimore) 83: 245-253. [Crossref]
- Modlin JF (1986) Perinatal echovirus infection: insights from a literature review of 61 cases of serious infection and 16 outbreaks in nurseries. Rev Infect Dis 8: 918-926. [Crossref]
- Bogomolov BP, Deviatkin AV, Mitiushina SA, Mol'kova TN (2007) Acute myocarditis caused by ECHO virus. Klin Med (Mosk) 85: 68-70. [Crossref]
- Midulla M, Marzetti G, Borra G, Sabatino G (1976) Myocarditis associated with Echo type 7 infection in a leukemic child. Acta Paediatr Scand 65: 649-651. [Crossref]
- Rupprecht CE (1996) Rhabdoviruses: rabies virus. Medical Microbiology. In Baron S. (Eds) Medical Microbiology. 4th edition. Galveston (TX): University of Texas Medical Branch at Galveston: Chapter 61. [Crossref]
- Davis BM, Rall GF, Schnell MJ (2015) Everything you always wanted to know about rabies virus (but were afraid to ask). Annu Rev Virol 2: 451-471. [Crossref]
- Ross E, Armentrout SA (1962) Myocarditis associated with rabies: report of a case. New Eng J Med 266: 1087-1089. [Crossref]
- Araujo MD, BRITO TD, Machado CG (1971) Myocarditis in human rabies. Rev Inst Med Trop Sao Paulo 13: 99-102. [Crossref]
- Cheetham HD, Hart J, Coghill NF, Fox B (1970) Rabies with myocarditis: two cases in England. Lancet 295: 921-922. [Crossref]
- Raman GV, Prosser A, Spreadbury PL, Cockcroft PM, Okubadejo OA (1988) Rabies presenting with myocarditis and encephalitis. J Infect 17: 155-158. [Crossref]
- Maniloff J, Das J, Christensen JR (1977) Viruses of mycoplasmas and spiroplasmas. Adv Virus Res 2: 343-380. [Crossref]
- Jansson E, Backman A, Hakkarainen K, Miettinen A (1982) Viruses of mycoplasmas and spiroplasmas. Med Biol 60: 125-131. [Crossref]
- Thacker EL, Halbur PG, Ross RF, Thanawongnuwech R, Thacker BJ (1999) Mycoplasma hyopneumoniae potentiation of porcine reproductive and respiratory syndrome virus-induced pneumonia. J Clin Microbiol 37: 620-627. [Crossref]
- Smyth A (2002) Pneumonia due to viral and atypical organisms and their sequelae: childhood respiratory infections. Br Med Bull 61: 247-262. [Crossreff]
- Formosa GM, Bailey M, Barbara C, Muscat C, Grech V (2006) Mycoplasma pneumonia–an unusual cause of acute myocarditis in childhood. Images Paediatr Cardiol 8:7-10. [Crossref]
- Lewes D, Rainford DJ, Lane WF (1974) Symptomless myocarditis and myalgia in viral and Mycoplasma pneumoniae infections. Br Heart J 36: 924-932. [Crossref]
- Meiklejohn G, Beck MD, Eaton MD (1944) Atypical pneumonia caused by psittacosis-like viruses. J Clin Invest 23: 167-175. [Crossref]
- Bedson SP, Bland JO (1934) The developmental forms of psittacosis virus. Br J Exp Pathol15: 243-247. [Crossref]
- Burijan J, Micic J (1957) Pneumonia caused by psittacosis virus (ornithosis virus). Srp Arh Celok Lek 85: 1260-1267. [Crossref]
- Weygaerde YV, Versteele C, Thijs E, De Spiegeleer A, Boelens J et al. (2018) An unusual presentation of a case of human psittacosis. Respir Med Case Rep 23: 138-142. [Crossref]
- Fauci AS (1988) The human immunodeficiency virus: infectivity and mechanisms of pathogenesis. Science 239: 617-622. [Crossref]
- Rerkpattanapipat P, Wongpraparut N, Jacobs LE, Kotler MN (2000) Cardiac manifestations of acquired immunodeficiency syndrome. Arch Intern Med 160: 602-608. [Crossref]
- Anderson DW, Virmani R, Reilly JM, O'Leary T, Cunnion RE et al. (1988) Prevalent myocarditis at necropsy in the acquired immunodeficiency syndrome. J Am Coll Cardiol 11: 792-799. [Crossref]
- Baroldi G, Corallo S, Moroni M, Repossini A, Mutinelli MR et al. (1988) Focal lymphocytic myocarditis in acquired immunodeficiency syndrome (AIDS): a correlative morphologic and clinical study in 26 consecutive fatal cases. J Am Coll Cardiol 12: 463-469. [Crossref]
- Akhras FA (1993) HIV and opportunistic infections: which makes the heart vulnerable? Br J Clin Pract 47: 232-233. [Crossref]
- Grody WW, Cheng L, Lewis W (1990) Infection of the heart by the human immunodeficiency virus. Am J Cardiol 66: 203-206. [Crossref]
- Acierno LJ (1989) Cardiac complications in acquired immunodeficiency syndrome (AIDS): a review. J Am Coll Cardiol 13: 1144-1154. [Crossref]
- Blanchard DG, Hagenhoff C, Chow LC, McCann HA, Dittrich HC (1991) Reversibility of cardiac abnormalities in human immunodeficiency virus (HIV)-infected individuals: a serial echocardiographic study. J Am Coll Cardiol 17: 1270-1276. [Crossref]
- Levy WS, Simon GL, Rios JC, Ross AM (1989) Prevalence of cardiac abnormalities in human immunodeficiency virus infection. Am J Cardiol 63: 86-89. [Crossref]
- Himelman RB, Chung WS, Chernoff DN, Schiller NB, Hollander H (1989) Cardiac manifestations of human immunodeficiency virus infection: a two-dimensional echocardiographic study. J Am Coll Cardiol 13: 1030-1036. [Crossref]
- Herskowitz A, Vlahov D, Willoughby S, Chaisson RE, Schulman SP et al. (1993) Prevalence and incidence of left ventricular dysfunction in patients with human immunodeficiency virus infection. Am J Cardiol 71: 955-958. [Crossref]
- Jacob AJ, Boon NA (1991) HIV cardiomyopathy: a dark cloud with a silver lining? Br Heart J 66: 1-2. [Crossref]
- Barbaro G, Di Lorenzo G, Soldini M, Giancaspro G, Grisorio B et al. (1999) Intensity of myocardial expression of inducible nitric oxide synthase influences the clinical course of human immunodeficiency virus-associated cardiomyopathy. Circulation 100: 933-939. [Crossref]
- Gould E, Pettersson J, Higgs S, Charrel R, de Lamballerie X (2017) Emerging arboviruses: why today? One Health 4: 1-3. [Crossref]
- Beckham JD, Tyler KL (2015) Arbovirus infections. Continuum (Minneap Minn 6 Neuroinfectious Disease) 1599-1611. [Crossref]
- Obeyesekere I, Hermon Y (1972) Myocarditis and cardiomyopathy after arbovirus infections (dengue and chikungunya fever). Br Heart J 34: 821-827. [Crossref]
- Nagaratnam N, Siripala K, de Silva N (1973) Arbovirus (dengue type) as a cause of acute myocarditis and pericarditis. Br Heart J 35: 204-206. [Crossref]
- Aletti M, Lecoules S, Kanczuga V, Soler C, Maquart M et al. (2017) Transient myocarditis associated with acute Zika virus infection. Clin Infect Dis 64: 678-679. [Crossref]
- Kawai C (1999) From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation 99: 1091-1100. [Crossref]
- Cooper Jr LT (2009) Myocarditis. N Engl J Med 360: 1526-1538. [Crossref]
- Kindermann I, Barth C, Mahfoud F, Ukena C, Lenski M et al. (2012) Update on myocarditis. J Am Coll Cardiol 59: 779-792. [Crossref]
- Krejci J, Mlejnek D, Sochorova D, Nemec P (2016) Inflammatory cardiomyopathy: a current view on the pathophysiology, diagnosis, and treatment. Biomed Res Int. 2016;2016. [Crossref]
- Dennert R, Crijns HJ, Heymans S (2008) Acute viral myocarditis. Eur Heart J 29: 2073-2082. [Crossref]
- Kuffner M, Pawlak A, Przybylski M (2016) Viral Infection of the Heart: Pathogenesis and Diagnosis. Pol J Microbiol 65: 391-398. [Crossref]
- Kearney MT, Cotton JM, Richardson PJ, Shah AM (2001) Viral myocarditis and dilated cardiomyopathy: mechanisms, manifestations, and management. Postgrad Med J. 7: 4-10. [Crossref]
- Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C et al. (2003) Interferon-β treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 107: 2793-2798. [Crossref]
- Shi Y, Chen C, Lisewski U, Wrackmeyer U, Radke M rt al. (2009) Cardiac deletion of the Coxsackievirus-adenovirus receptor abolishes Coxsackievirus B3 infection and prevents myocarditis in vivo J Am Coll Cardiol 53: 1219-1226. [Crossref]
- Noutsias M, Pauschinger M, Poller WC, Schultheiss HP, Kuhl U (2003) Current insights into the pathogenesis, diagnosis and therapy of inflammatory cardiomyopathy. Heart Fail 3: 127-135. [Crossref]
- Martino TA, Petric M, Brown M, Aitken K, Gauntt CJ et al. (1998) Cardiovirulent coxsackieviruses and the decay-accelerating factor (CD55) receptor. Virology 244: 302-314. [Crossref]
- Tracy S, Höfling K, Pirruccello S, Lane PH, Reyna SM et al. (2000) Group B coxsackievirus myocarditis and pancreatitis: connection between viral virulence phenotypes in mice. J Med Virol 62: 70-81. [Crossref]
- Beck MA, Shi Q, Morris VC, Levander OA (1995) Rapid genomic evolution of a non-virulent coxsackievirus B3 in selenium-deficient mice results in selection of identical virulent isolates. Nat Med 1: 433-436. [Crossref]
- Ilback NG, Wesslén L, Fohlman J, Friman G (1996) Effects of methyl mercury on cytokines, inflammation and virus clearance in a common infection (coxsackie B3 myocarditis). Toxicology Letters 89: 19-28. [Crossref]
- Cooper LT, Rader V, Ralston NV (2007) The roles of selenium and mercury in the pathogenesis of viral cardiomyopathy. Cong Heart Fail 13: 193-199. [Crossref]
- McManus BM, Chow LH, Wilson JE, Anderson DR, Gulizia JM et al. (1993) Direct myocardial injury by enterovirus: a central role in the evolution of murine myocarditis. Clin Immunol Immunopathol 68: 159-169. [Crossref]
- Maekawa Y, Ouzounian M, Opavsky MA, Liu PP (2007) Connecting the missing link between dilated cardiomyopathy and viral myocarditis: virus, cytoskeleton, and innate immunity. Circulation 115: 5-8. [Crossref]
- Matzinger P (2002) The danger model: a renewed sense of self. Science 296: 301-305. [Crossref]
- Sagar S, Liu PP, Cooper Jr LT (2012) Myocarditis. Lancet 379: 738-747. [Crossref]
- Yajima T (2011) Viral myocarditis: potential defense mechanisms within the cardiomyocyte against virus infection. Future Microbiol 6: 551-566. [Crossref]
- McCarthy RE, Boehmer JP, Hruban RH, Hutchins GM, Kasper EK et al. (2000) Long-term outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis. N Engl J Med 342: 690-695. [Crossref]
- Woodruff JF, Woodruff JJ (1974). Involvement of T lymphocytes in the pathogenesis of coxsackie virus B3 heart disease. J Immunol 113: 1726-1734. [Crossref]
- Fairweather D, Frisancho-Kiss S, Yusung S, Barrett M, Gatewood S et al. (2004) IFN-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines TGF-beta 1 and IL-4. Am J Pathol 165: 1883-1894. [Crossref]
- Maisch B, Pankuweit S (2013) Standard and etiology-directed evidence-based therapies in myocarditis: state of the art and future perspectives. Heart Fail Rev 18: 761-795. [Crossref]
- Li K, Xu W, Guo Q, Jiang Z, Wang P et al. (2009) Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Cir Res 105: 353-364. [Crossref]
- Pankuweit S, Moll R, Baandrup U, Portig I, Hufnagel G et al. (2003) Prevalence of the parvovirus B19 genome in endomyocardial biopsy specimens. Human Pathol 34: 497-503. [Crossref]
- Caforio AL, Mahon NJ, Tona F, McKenna WJ (2002) Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur J Heart Fail 4: 411-417. [Crossref]
- Schulze K, Becker BF, Schauer R, Schultheiss HP (1990) Antibodies to ADP-ATP carrier--an autoantigen in myocarditis and dilated cardiomyopathy--impair cardiac function. Circulation 81: 959-969. [Crossref]
- Li Y, Heuser JS, Cunningham LC, Kosanke SD, Cunningham MW (2006) Mimicry and antibody-mediated cell signaling in autoimmune myocarditis. J Immunol 177: 8234-8240. [Crossref]
- Galvin JE, Hemric ME, Ward K, Cunningham MW (2000) Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J Clin Invest 106: 217-224. [Crossref]
- Antone SM, Adderson EE, Mertens NM, Cunningham MW (1997) Molecular analysis of V gene sequences encoding cytotoxic anti-streptococcal/anti-myosin monoclonal antibody 36.2. 2 that recognizes the heart cell surface protein laminin. J Immunol 159: 5422-5430. [Crossref]
- Huber SA, Budd RC, Rossner K, Newell MK (1999) Apoptosis in coxsackievirus B3‐induced myocarditis and dilated cardiomyopathy. Ann N Y Acad Sci 887: 181-190. [Crossref]
- Kawai C, Matsumori A (2013) Dilated cardiomyopathy update: infectious-immune theory revisited. Heart Fail Rev 18: 703-714. [Crossref]
- Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP et al (1999) Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med 5: 320-326. [Crossref]
- Badorff C, Knowlton KU (2004) Dystrophin disruption in enterovirus-induced myocarditis and dilated cardiomyopathy: from bench to bedside. Med Microbiol Immunol 193: 121-126. [Crossref]
- Kim KS, Tracy S, Tapprich W, Bailey J, Lee CK et al. (2005) 5-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J Virol 79: 7024-7041. [Crossref]
- Li Y, Bourlet T, Andreoletti L, Mosnier JF, Peng T et al. (2000) Enteroviral capsid protein VP1 is present in myocardial tissues from some patients with myocarditis or dilated cardiomyopathy. Circulation 101: 231-234. [Crossref]
- Bultmann BD, Klingel K, Sotlar K, Bock CT, Baba HA et al. (2003) Fatal parvovirus B19–associated myocarditis clinically mimicking ischemic heart disease: an endothelial cell–mediated disease. Human Pathol 34: 92-95. [Crossref]
- Bock CT, Klingel K, Aberle S, Duechting A, Lupescu A et al. (2005) Human parvovirus B19: a new emerging pathogen of inflammatory cardiomyopathy. J Vet 52: 340-343. [Crossref]
- Klingel K, Sauter M, Bock CT, Szalay G, Schnorr JJ et al. (2014) Molecular pathology of inflammatory cardiomyopathy. Med Microbiol Immunol 193: 101-117. [Crossref]
- Krueger GR, Rojo J, Buja LM, Lassner D, Kuehl U (2008) Human herpesvirus-6 (HHV-6) is a possible cardiac pathogen: an immunopathological and ultrastructural study. Hosp Gen 71: 187-191.
- Schmidt-Lucke C, Spillmann F, Bock T, Kühl U, Van Linthout S et al. (2010) Interferon beta modulates endothelial damage in patients with cardiac persistence of human parvovirus B19 infection. J Infect Dis 201: 936-945. [Crossref]
- Brown KE, Anderson SM, Young NS (1993) Erythrocyte P antigen: cellular receptor for B19 parvovirus. Science 262: 114-117. [Crossref]
- Weigel-Kelley KA, Yoder MC, Srivastava A (2003) α5β1 integrin as a cellular coreceptor for human parvovirus B19: requirement of functional activation of β1 integrin for viral entry. Blood 102: 3927-3933. [Crossref]
- Munakata Y, Saito-Ito T, Kumura-Ishii K, Huang J, Kodera T et al. (2005) Ku80 autoantigen as a cellular co-receptor for human parvovirus B19 infection. Blood 106: 3449-3456. [Crossref]
- Moffatt S, Yaegashi N, Tada K, Tanaka N, Sugamura K (1998) Human parvovirus B19 nonstructural (NS1) protein induces apoptosis in erythroid lineage cells. J Virol 72: 3018-3028. [Crossref]
- Hsu TC, Tzang BS, Huang CN, Lee YJ, Liu GY et al. (2006) Increased expression and secretion of interleukin‐6 in human parvovirus B19 non‐structural protein (NS1) transfected COS‐7 epithelial cells. Clin Exp Immunol 144: 152-157. [Crossref]
- Fu Y, Ishii KK, Munakata Y, Saitoh T, Kaku M et al. (2002) Regulation of tumour necrosis factor alpha promoter by human parvovirus B19 NS1 through activation of AP-1 and AP-2. J Virol 76: 5395-5403. [Crossref]
- Duechting A, Tschöpe C, Kaiser H, Lamkemeyer T, Tanaka N et al. (2008) Human parvovirus B19 NS1 protein modulates inflammatory signaling by activation of STAT3/PIAS3 in human endothelial cells. J Virol 82: 7942-7952. [Crossref]
- Poole BD, Karetnyi YV, Naides SJ (2004) Parvovirus B19-induced apoptosis of hepatocytes. J Virol 78: 7775-7783. [Crossref]
- Poole BD, Zhou J, Grote A, Schiffenbauer A, Naides SJ (2006) Apoptosis of liver-derived cells induced by parvovirus B19 nonstructural protein. J Virol 80: 4114-4121. [Crossref]
- Schultheiss HP, Kühl U, Cooper LT (2011) The management of myocarditis. Eur Heart J 32: 2616-2625. [Crossref]
- Rotola A, Di Luca D, Cassai E, Ricotta D, Giulio A, Turano A, Caruso A, Muneretto C. Human herpesvirus 6 infects and replicates in aortic endothelium. J Clin Microbiol 38: 3135-3136. [Crossref]
- Wu CA, Shanley JD (1998) Chronic infection of human umbilical vein endothelial cells by human herpesvirus-6. J Gen Virol 79: 1247-1256. [Crossref]
- Caruso A, Rotola A, Comar M, Favilli F, Galvan M et al. (2002). HHV‐6 infects human aortic and heart microvascular endothelial cells, increasing their ability to secrete proinflammatory chemokines J Med Virol 67: 528-533. [Crossref]
- Takatsuka H, Wakae T, Mori A, Okada M, Fujimori Y et al. (2003) Endothelial damage caused by cytomegalovirus and human herpesvirus-6. Bone Marrow 31: 475-479. [Crossref]
- Vallbracht KB, Schwimmbeck PL, Kühl U, Seeberg B, Schultheiss HP (2004) Endothelium-dependent flow-mediated vasodilation of systemic arteries is impaired in patients with myocardial virus persistence. Circulation 110: 2938-2945. [Crossref]
- Tschope C, Bock CT, Kasner M, Noutsias M, Westermann D et al. (2005) High prevalence of cardiac parvovirus B19 infection in patients with isolated left ventricular diastolic dysfunction. Circulation 111: 879-886. [Crossref]
- Fischer D, Rossa S, Landmesser U, Spiekermann S, Engberding N et al. (2005) Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur Heart J 26: 65-69. [Crossref]
- Yilmaz A, Kindermann I, Kindermann M, Mahfoud F, Ukena C et al. (2010) Comparative evaluation of left and right ventricular endomyocardial biopsy. Circulation 122: 900-909. [Crossref]
- Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J et al. (2013) Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 34: 2636-2648. [Crossref]
- Maisch B, Pankuweit S (2012) Current treatment options in (peri) myocarditis and inflammatory cardiomyopathy. Herz 37: 644-656. [Crossref]
- Krejci J, Hude P, Poloczkova H, Zampachova V, Stepanova R et al. (2016) Correlations of the changes in bioptic findings with echocardiographic, clinical and laboratory parameters in patients with inflammatory cardiomyopathy. Heart and Vessels 31: 416-426. [Crossref]
- Ukena C, Mahfoud F, Kindermann I, Kandolf R, Kindermann M et al. (2011) Prognostic electrocardiographic parameters in patients with suspected myocarditis. Eur J Heart Fail 13: 398-405. [Crossref]
- Morgera T, Di Lenarda A, Dreas L, Pinamonti B, Humar F et al. (1992) Electrocardiography of myocarditis revisited: clinical and prognostic significance of electrocardiographic changes. Am Heart J 124: 455-467. [Crossref]
- Vignola PA, Aonuma K, Swaye PS, Rozanski JJ, Blankstein RL et al. (1984) Lymphocytic myocarditis presenting as unexplained ventricular arrhythmias: diagnosis with endomyocardial biopsy and response to immunosuppression. J Am Coll Cardiol 4: 812-819. [Crossref]
- Dec GW, Waldman H, Southern J, Fallon JT, Hutter AM et al. (1992) Viral myocarditis mimicking acute myocardial infarction. J Am Coll Cardiol 20: 85-89. [Crossref]
- Magnani JW, Danik HJ, Dec Jr GW, DiSalvo TG (2006) Survival in biopsy-proven myocarditis: a long-term retrospective analysis of the histopathologic, clinical, and hemodynamic predictors. Am Heart J 151: 463-470. [Crossref]
- Nakashima H, Katayama T, Ishizaki M, Takeno M, Honda Y et al. (1998) Q wave and non-Q wave myocarditis with special reference to clinical significance. Jpn Heart J 39: 763-774. [Crossref]
- Greenwood RD, Nadas AS, Fyler D (1976) The clinical course of primary myocardial disease in infants and children. Am Heart J 92: 549-560. [Crossref]
- Pinamonti B, Alberti E, Cigalotto A, Dreas L, Salvi A et al. (1988) Echocardiographic findings in myocarditis. Am J Cardiol 62: 285-291. [Crossref]
- Escher F, Westermann D, Gaub R, Pronk J, Bock T et al. (2011) Development of diastolic heart failure in a 6-year follow-up study in patients after acute myocarditis. Heart 97: 709-714. [Crossref]
- Angelini A, Calzolari V, Calabrese F, Boffa GM, Maddalena F et al. (2000) Myocarditis mimicking acute myocardial infarction: Role of endomyocardial biopsy in the differential diagnosis. Heart 84: 245-250. [Crossref]
- Felker GM, Boehmer JP, Hruban RH, Hutchins GM, Kasper EK et al. (2000) Echocardiographic findings in fulminant and acute myocarditis. J Am Coll Cardiol 36: 227-232. [Crossref]
- Mendes LA, Dec GW, Picard MH, Palacios IF, Newell J et al. (1994) Right ventricular dysfunction: An independent predictor of adverse outcome in patients with myocarditis. Am Heart J 128: 301-307. [Crossref]
- Friedrich MG, Sechtem U, Schulz-Menger J, Holmvang G, Alakija P et al. (2009) Cardiovascular magnetic resonance in myocarditis: A JACC White Paper. J Am Coll Cardiol 53: 1475-1487. [Crossref]
- Baccouche H, Mahrholdt H, Meinhardt G, Merher R, Voehringer M et al. (2009) Diagnostic synergy of non-invasive cardiovascular magnetic resonance and invasive endomyocardial biopsy in troponin-positive patients without coronary artery disease. Eur Heart J 30: 2869-2879. [Crossref]
- Holzmann M, Nicko A, Kühl U, Noutsias M, Poller W et al. (2008) Complication rate in right ventricular endomyocardial biopsy–A retro-and prospective study over a 11 year period analyzing 3048 diagnostic procedures. Circulation 118: 1722-1728.
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE et al. (2013) 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol 62: 1495-539. [Crossref]
- Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M et al. (2007) The role of endomyocardial biopsy in the management of cardiovascular disease: A scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology J Am Coll Cardiol 50: 1914-1931. [Crossref]
- Frustaci A, Russo MA, Chimenti C (2009) Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J 30: 1995-2002. [Crossref]
- Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG et al. (2016) 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 18: 891-975. [Crossref]
- Rezkalla SH, Raikar S, Kloner RA (1996) Treatment of viral myocarditis with focus on captopril. Am J Cardiol 77: 634-637. [Crossref]
- Saegusa S, Fei Y, Takahashi T, Sumino H, Moriya J et al. (2007) Oral administration of candesartan improves the survival of mice with viral myocarditis through modification of cardiac adiponectin expression. Cardiovasc Drugs Ther 21: 155-160. [Crossref]
- Costanzo-Nordin MR, Reap EA, O'connell JB, Robinson JA, Scanlon PJ (1985) A nonsteroid anti-inflammatory drug exacerbates Coxsackie B3 murine myocarditis. Cardiology. J Am Coll Cardiol 6: 1078-1082. [Crossref]
- Schultheiss HP, Kuehl U (2010) Cardiovascular viral infections. In: Lennette EH (ed), Lennette’s Laboratory Diagnosis of Viral Infections, Chapter 18. 4th Ed. New York: Informa Healthcare USA Inc. pp. 304-318.
- Juhl U, Strauer BE, Schultheiss HP (1994) Methylprednisolone in chronic myocarditis. Postgrad Med J 70: S35-S42. [Crossref]
- Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, Glanowska G, Wilczewski P et al. (2001) Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two-year follow-up results. Circulation 104: 39-45. [Crossref]
- Noutsias M, Seeberg B, Schultheiss HP, Kühl U (1999) Expression of cell adhesion molecules in dilated cardiomyopathy: evidence for endothelial activation in inflammatory cardiomyopathy. Circulation 99: 2124-2131. [Crossref]
- Drucker NA, Colan SD, Lewis AB, Beiser AS, Wessel DL et al. (1994) Gamma-globulin treatment of acute myocarditis in the paediatric population. Circulation 89: 252-257. [Crossref]
- English RF, Janosky JE, Ettedgui JA, Webber SA (2004) Outcomes for children with acute myocarditis. Cardiol Young 14: 488-493. [Crossref]
- Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK et al. (2009) A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol 54: 2277-2286. [Crossref]
- Liu C, Chen J, Liu K (2005) Immunosuppressive treatment for inflammatory cardiomyopathy. Int Heart J 46: 113-122. [Crossref]
- Hia CP, Yip WC, Tai BC, Quek SC (2004) Immunosuppressive therapy in acute myocarditis: an 18 year systematic review. Arch Dis Child 89: 580-584. [Crossref]
- Winter MP, Sulzgruber P, Koller L, Bartko P, Goliasch G et al. (2018) Immunomodulatory treatment for lymphocytic myocarditis: a systematic review and meta-analysis. Heart Fail Rev 23: 573-581. [Crossref]
- Stanton C, Mookadam F, Cha S, McNamara D, Aukrust P et al. (2008) Greater symptom duration predicts response to immunomodulatory therapy in dilated cardiomyopathy. Int J Cardiol 128: 38-41. [Crossref]
- Moher D, Liberati A, Tetzlaff J, Altman DG, Prisma Group (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Medicine 6: e1000097. [Crossref]
- Parrillo JE, Cunnion RE, Epstein SE, Parker MM, Suffredini AF et al. (1989) A prospective, randomized, controlled trial of prednisone for dilated cardiomyopathy. New Eng J Med 321: 1061-1068. [Crossref]
- Latham RD, Mulrow JP, Virmani R, Robinowitz M, Moody JM (1989) Recently diagnosed idiopathic dilated cardiomyopathy: incidence of myocarditis and efficacy of prednisone therapy. Am Heart J 117: 876-882. [Crossref]
- Chan KY, Iwahara M, Benson LN, Wilson GJ, Freedom RM (1991) Immunosuppressive therapy in the management of acute myocarditis in children: a clinical trial. J Am Coll Cardiol 17: 458-460. [Crossref]
- Drucker NA, Colan SD, Lewis AB, Beiser AS, Wessel DL et al. (1994) Gamma-globulin treatment of acute myocarditis in the pediatric population. Circulation 89: 252-257. [Crossref]
- Balaji S, Wiles HB, Sens MA, Gillette PC (1994) Immunosuppressive treatment for myocarditis and borderline myocarditis in children with ventricular ectopic rhythm. Heart 72: 354-359. [Crossref]
- Camargo PR, Snitcowsky R, Da Luz PL, Mazzieri R, Higuchi ML et al. (1995) Favorable effects of immunosuppressive therapy in children with dilated cardiomyopathy and active myocarditis. Pediatr Cardiol 16: 61-68. [Crossref]
- Mason JW, O'connell JB, Herskowitz A, Rose NR, McManus BM et al. (1995) Myocarditis treatment trial investigators. A clinical trial of immunosuppressive therapy for myocarditis. New Eng J Med 333: 269-275. [Crossref]
- Lee KJ, McCrindle BW, Bohn DJ, Wilson GJ, Taylor GP et al. (1999) Clinical outcomes of acute myocarditis in childhood. Heart 82: 226-233. [Crossref]
- Ahdoot J, Galindo A, Alejos JC, George B, Burch C et al. (2000) Use of OKT3 for acute myocarditis in infants and children. J Heart Lung Transplant 19: 1118-1121. [Crossref]
- Gullestad L, Aass H, Fjeld JG, Wikeby L, Andreassen AK et al. (2001) Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation 103: 220-225. [Crossref]
- McNamara DM, Holubkov R, Starling RC, Dec GW, Loh E et al. (2001) Controlled trial of intravenous immune globulin in recent-onset dilated cardiomyopathy. Circulation 103: 2254-2259. [Crossref]
- Staudt A, Schaper F, Stangl V, Plagemann A, Bohm M et al. (2001) Immunohistological changes in dilated cardiomyopathy induced by immunoadsorption therapy and subsequent immunoglobulin substitution. Circulation 103: 2681-2686. [Crossref]
- Frustaci A, Chimenti C, Calabrese F, Pieroni M, Thiene G et al. (2003) Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation 107: 857-863. [Crossref]
- English RF, Janosky JE, Ettedgui JA, Webber SA (2004) Outcomes for children with acute myocarditis. Cardiol Young 14: 488-493. [Crossref]
- Wojnicz R, Wilczek K, Nowalany-Kozielska E, Szyguła-Jurkiewicz B, Nowak J et al. (2006) Usefulness of atorvastatin in patients with heart failure due to inflammatory dilated cardiomyopathy and elevated cholesterol levels. Am J Cardiol 97: 899-904. [Crossref]
- Klugman D, Berger JT, Sable CA, He J, Khandelwal SG, et al. (2010) Pediatric patients hospitalized with myocarditis: a multi-institutional analysis. Pediatr Cardiol 31: 222-228. [Crossref]
- Escher F, Kühl U, Lassner D, Poller W, Westermann D et al. (2016) Long-term outcome of patients with virus-negative chronic myocarditis or inflammatory cardiomyopathy after immunosuppressive therapy. Clin Res Cardiol 105: 1011-1120. [Crossref]
- Merken J, Hazebroek M, Van Paassen P, Verdonschot J, Van Empel V et al. (2018) Immunosuppressive therapy improves both short-and long-term prognosis in patients with virus-negative nonfulminant inflammatory cardiomyopathy. Circulation Heart Fail 11: e004228. [Crossref]
- Aretz HT (1987) Myocarditis: the Dallas criteria. Hum Pathol 18: 619-624. [Crossref]
- Baughman KL (2006) Diagnosis of myocarditis: death of Dallas criteria. Circulation 113: 593-595. [Crossref]
- Mahrholdt H, Goedecke C, Wagner A, Meinhardt G, Athanasiadis A et al. (2004) Cardiovascular magnetic resonance assessment of human myocarditis: a comparison to histology and molecular pathology. Circulation 109: 1250-1258. [Crossref]
- Gutberlet M, Spors B, Thoma T, Bertram H, Denecke T et al. (2008) Suspected chronic myocarditis at cardiac MR: diagnostic accuracy and association with immunohistologically detected inflammation and viral persistence. Radiology 246: 401-409. [Crossref]
- Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M et al. (2007) The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. J Am Coll Cardiol 50: 1914-1931. [Crossref]
- Cooper LT (2009) The heat is off: immunosuppression for myocarditis revisited. Eur Heart J 30: 1936-1939. [Crossref]