The Zika virus (ZIKV) is a tropical and subtropical emergent pathogen, with main clinical manifestations of low fever, headache, myalgia, arthralgia in the small joints of the hands and feet, non-purulent conjunctivitis, ocular pain, prostration, and pruritic maculopapular rash. Furthermore, the most feared complication of this viral infection is microcephaly, caused by the interaction between ZIKV and cells from the fetal central nervous system (CNS). Identifying the mechanism and factors linked to the entry of ZIKV into human cells, particularly in the fetus during the first developmental months, is currently the greatest challenge in understanding the tropism and pathogenesis of ZIKV. Thus, this review aims to assess the ZIKV–human molecular interaction, the main cellular receptors involved in the virus and host, the viral infection process, and microcephaly neuropathogenesis. During ZIKV–human host interaction, the virus binds to host cell membrane receptors, followed by internalization (through endocytic vesicles) and inhibition of the innate immune response, similar to the normal process of receptor signaling activation. Infection of human fetuses by ZIKV leads to cell cycle deregulation, activating cell death by apoptosis, and microcephaly. Blocking the interaction between the virus and specific membrane receptors may be a good strategy to prevent ZIKV infection, particularly in pregnant women during the first months of fetal development. Thus, knowledge of the whole ZIKV–host interaction process may help in designing novel therapies or targets for drugs to prevent the death of fetal CNS cells and microcephaly.
zika virus, microcephaly, neuropathogenesis, host interaction, cell membrane receptors, arboviruses
Human actions, which alter the urban environment, greatly contribute to the problem of viruses because of demographic transition. This ecological modification is linked to vector transmission, particularly in tropical countries [1]. Such viruses are defined under the nosological category, which refers to diseases caused by viruses that propagate in the population, for example dengue, chikungunya, and Zika [2]. In Brazil in recent decades, there have been four main epidemiologically arboviruses, namely, chikungunya, dengue, yellow fever, and Zika, transmitted by the same vector, Aedes aegypti. The inability to contain mosquitoes increases their capacity for migration, making the human population more likely to encounter these diseases. Common clinical manifestations caused by these arboviruses are fever, myalgia, arthralgia, rashes, and headaches [3-6]. The primary symptom of chikungunya virus (CHIKV) is polyarthralgia, which is often accompanied by joint edema, differentiating it from the symptoms caused by other arboviruses, with an inflammatory response that may progress into subacute and chronic forms. Dengue can be divided into three clinical phases: febrile, critical, and recovery. The main aggravating factor of this condition is shock, hemorrhage, and organ dysfunction [7]. Different from these aforementioned diseases, the symptoms of yellow fever are varied, ranging from icterus, albuminuria, and hemorrhages as well as the Faget sign (an increase in body temperature with a slow pulse) [8]. Zika virus (ZIKV) infection may present with asymptomatic or oligosymptomatic signs that may be resolved in 2-7 days [7]. Symptoms of these patients are usually low fever (37.8-38.5 °C), headache, myalgia, arthralgia in the small joints of the hands and feet, nonpurulent conjunctivitis, ocular pain, prostration, and pruritic maculopapular rash [9-12].
ZIKV epidemiology
ZIKV belongs to the genus Flavivirus in the family Flaviviridae. This virus was discovered in 1947 in the blood from a feverish Rhesus monkey in the Zika forest near Lake Victoria on the outskirts of Entebbe, the capital of Uganda. It remained dormant for nearly six decades and was restricted to Africa and Asia, where the disease disseminated in a mild form [13,14]. The first human case was reported in 1954 [15]. The pandemic occurred mostly in tropical and subtropical areas, primarily in Africa, Asia, and the equatorial zone, and was restricted until 2007. A large-scale infection was reported in Micronesia, and in 2013–2014, an epidemic occurred in French Polynesia and New Caledonia. Furthermore, it has expanded to South and Central America and the Caribbean Islands. In Latin America, Brazil was one of the countries the most affected by ZIKV, wherein the first reported case occurred in 2015 [11,12,16-18]. The spread of ZIKV was influenced by climatic and geographic dynamics, globalization, and particularly by unreliable sanitation and poorly planned urbanization that facilitated mosquito resistance and spread of disease in more advanced stages. This has garnered the attention of researchers on the need for further studies in the light of this epidemic and research for the development of an effective vaccine [13,14]. In Brazil, the main states affected were Pernambuco, Bahia, and Paraíba, as these states had the contributing factors required for the proliferation of the virus and vector, such as global warming and climatic changes in association with the El Niño climate phenomenon, socioeconomic conditions, and a lack of environmental awareness by the population in these regions [9,11]. The spread of this virus was primarily due to the Olympic Games (2016) and World Cup Soccer (2014) that occurred in Brazil, thus, raising the interest of researchers. Therefore, the World Health Organization (WHO) declared a public health emergency of international concern [19]. Initially, there was a suspicion that ZIKV could be one of the causes of Guillain–Barré syndrome (GBS); therefore, case–control studies were performed to determine whether there was a correlation between these pathologies. A survey was conducted between October 2013 and April 2014 in French Polynesia during an outbreak of ZIKV, and an increase in the incidence of GBS was noticed. In the case group, there were 98 subjects, of which 42 were diagnosed with GBS and 41 tested positive for anti-Zika IgM or IgG antibody. From the 42 patients with GBS, 39 had ZIKV IgM (i.e., recent infection by ZIKV) and 37 presented with a transient clinical condition an average of six days before the initiation of neurological symptoms. The mean duration of the disease was six days, and the plateau was four days. With this study, it is evident that ZIKV infection is one of the causes of GBS, thus, placing more emphasis on the need for intensive therapies to minimize pathological damages [20].
ZIKV and microcephaly
In November 2015 in Brazil, a specific notification system for term infants (gestational age of ≥37 weeks) with a small head circumference (≤32 cm in diameter) was introduced following a microcephaly outbreak in the northeastern region of Brazil [21,22]. However, it was only in March 2016 that WHO adopted the criteria for cephalic perimeter of 31.7 cm in boys and 31.5 cm in girls. ZIKV infection was declared as a public health emergency of international interest owing to serious clinical manifestations, including fetal anomalies, neurological problems, and autoimmune disorders [23]. With the birth of children with microcephaly in Brazil, in October 2015, the Pan American Health Organization (PAHO) began an investigation in the city of Recife in the northeastern region of Brazil. Imaging tests, such as tomography and ultrasound, revealed a small forehead, reduction in the space between the bones of the skull, developmental defects of the brain, enlarged cerebral ventricles, and various calcifications. These characteristics closely resembled congenital symptoms caused by cytomegalovirus or rubella [24]. In the same study, the mothers of children with microcephaly reported having had a mild disease in early gestation, presenting with rashes, skin patches, and fever. To exclude differential diagnoses, these mothers were investigated by analyzing their habits and possible contact with medications, foods, illicit drug use, and environmental pollutants. During this investigation, none of the suspected reasons was confirmed, except for microencephalic expansion in the newborns in the state of Pernambuco. Subsequently, the same pathological evidence was observed in other states of Brazilian northeast region. Thus, WHO declared a need for national research to broaden the diagnosis of the disease [24]. The Brazilian research institution Oswaldo Cruz (Fiocruz) in Paraíba state detected ZIKV in the amniotic fluid of pregnant women in November 2015 [25]. In December of the same year, PAHO detected the presence of viral RNA using the RT-PCR approach in the amniotic fluid samples of two pregnant women who had their fetuses diagnosed with microcephaly (at the prenatal ultrasound imaging), and one of these children died during the neonatal period [6]. Analyses were also carried out in French Polynesia because, similar to those in Brazil, there were a number of children born with microcephaly and other malformations of the CNS. In French Polynesia, studies were performed from March 2014 until May 2015, but only non-specific samples were identified, i.e., IgG antibody positivity only for flavivirus, in asymptomatic pregnant women [13,26]. Based on these findings, Brazilian researchers, in conjunction with the re-evaluation of research from French Polynesia, identified cases of severe fetal neurological problems that occurred between 2013 and 2014 that were also associated with ZIKV [27,28].
ZIKV-molecular structure
The ZIKV genome is a single-stranded positive RNA, with sequencing of a specific viral strain indicating that it was 10,806 bp [29]. This RNA encodes for structural and non-structural proteins (Figure 1). The structural proteins found in ZIKV are called C (capsid), prM (pre-membrane), and E (envelope) and are responsible for forming the capsid and envelope of the virus [30-32]. Protein E is the major target of neutralizing antibodies and contains three domains (I, II, and III); it is an envelope of the surface glycoprotein of the virus and functions as a fusion protein between the virus and host membrane receptors [31,33-35]. Protein E acts on different critical viral life cycle mechanisms, such as coupling with the host cell, binding and entry factors, cell surface receptors, and membrane fusion, and these factors determine the cellular tropism of the virus. Moreover, protein E is related to the process of virus assembly [32,36]. The prM protein (precursor membrane protein) is, as its name suggests, the precursor of the M (matrix) protein (Figure 1). The prM/M complex protects protein E from degradation during virion assembly [31,35].
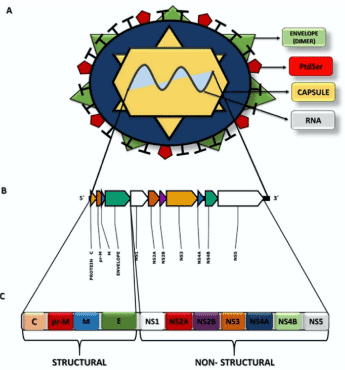
Figure 1. Structural representation of ZIKV. A Viral structures and their distribution for formation of the virion and its genome. B The ZIKV RNA molecule and distribution of the genes for formation of the viral proteins. C Representation of the polyprotein that will be cleaved to form viral proteins (structural and non-structural) from viral RNA
Domain I is the N-terminal region of the E protein, which is related to the immune response of non-neutralizing antibodies, i.e., it cannot prevent the proliferation of antigens. Thus, mutations in these domains allow for increased virulence, infectivity, and host–virus tropism [33,36]. In Brazil, it was verified that ZIKV belongs to the Asian strains but with some alterations in the E protein sequence. As a consequence of these mutations, there are two substitutions in the amino acid sequence, one at position 279, wherein phenylalanine (Phe) is replaced with serine (Ser), located at the fusion point between the domains I and II. This position is hydrophobic and allows alterations in the arrangement of viral proteins induced by low pH, enabling the fusion of the virus with the host cell [33,37,38]. The second mutation is a change in isoleucine (Ile) to valine (Val) at position 311 in domain III of the E protein. This alteration in the protein primary structure enabled the changing of the polarity and increase in polar site, thus, enabling the virus to neutralize the IgM immunoglobulin. Prior to this modification, this domain was the viral epitope [33,38].
Among the non-structural proteins (NS), NS1 is present in the early stages of viral replication. NS1 regulates the functions of the viral replication cycle, including control of synthesis, splicing, transport, and translation of mRNAs. In addition, NS1 is secreted into the extracellular medium in the serum of patients during the acute phase of infection [39,40]. The NS2A protein comprises the viral replication complex that acts on the formation of the virus and blocks the host immune response [40]. NS2B is a cofactor for the NS3 protease that cleaves the protein junctions in the viral polyprotein and is fundamental for replication. The NS3 protein exhibits multiple enzymatic activities, viz., helicase, which is energy dependent, serine protease, and RNA triphosphatase. NS4A binds viral replication to the cell membrane and induces autophagy in the infected cells, favoring replication. NS4B assists NS3 in viral replication. NS5 is a protein that has several enzymatic activities for viral replication. In the N-terminal region, there are guanylyltransferase and S-adenosylmethionine-dependent methyltransferase domains, which are essential for the methylation of the 5′-end of the viral RNA (cap 5′). The C-terminal region has RNA-dependent RNA polymerase function, which acts on the synthesis of RNA of negative (replicative) and positive polarity [30,39].
Molecular interaction of ZIKV with human host
During the interaction of ZIKV with the human host, the first cells the virus encounters are dermal fibroblasts, epidermal keratinocytes, and immature dendritic cells. From which here it is transmitted to the dermal dendritic cells (Langerhans cells), facilitating the dispersion of ZIKV. As described for dengue virus (DENV), ZIKV appears to induce cell death by apoptosis in epidermal cells from infected epithelial tissues, a mechanism capable of evading the immune system, thus, increasing the viral load [41].
Several putative host membrane receptors used by flaviviruses (primarily DENV) have been described in both mammalian and mosquito cells [32,41-44]. The most well-characterized protein families, which flaviviruses bind to for initiating infection, are C-type lectin receptors (CLRs), phosphatidylserine receptors (PtdSer), T-cell immunoglobulin and mucin domain (TIM) receptors, and TAM family tyrosine kinase receptors (TYRO3, AXL, and MER). CLRs are specialized to detect invading pathogens and play a central role in the activation of host immune defenses. TIM and TAM receptors participate in the process of phagocytosis (dependent on phosphatidylserine) and in the elimination of apoptotic cells. In addition, TAM receptors interact with multiple signaling molecules to regulate cell migration, survival, and the clearance of metabolic products and cellular debris. TAM receptors also reduce the expression of pro-inflammatory molecules [45]. Studies have indicated that ZIKV entry into fibroblasts, keratinocytes, and human immature dendritic cells is mediated by specific receptors, such as DC-SIGN (dendritic cell-specific intracellular adhesion molecule-3-grabbing non-integrin, a specific type of CLR), AXL, TYRO3, and, to a lesser extent, TIM-1. In addition, TLR3 (Toll-like receptor 3), a specific receptor pattern recognition receptor (PRR), capable of detecting the presence of pathogen-associated molecular patterns (PAMPs), is the initial immune receptor during the detection of ZIKV infection in human fibroblasts, thereby stimulating immune responses by the production of type I and type II interferons (IFN) [32,41-44].
ZIKV in fetal vertical transmission
It is possible to detect the presence of ZIKV in placental chorionic villi, fetal tissues, intervillous space, the brain, and amniotic fluid [39,46-49]. The placenta is a physical and immunological barrier consisting of primary human trophoblasts (PHTs), which are composed of cytotrophoblast and syncytiotrophoblast cells. Placental PHTs make it difficult for the virus to contact the fetus due to the production of antiviral IFNλ1, acting in an autocrine and paracrine manner to protect PHTs and non-trophoblast cells [32,50]. Thus, for syncytiotrophoblast infection, ZIKV must avoid inhibition by IFNλ1 and other antivirals produced by trophoblasts. However, for ZIKV to cross the barrier in the second half of pregnancy, it is necessary to consider alternative methods, such as non-trophoblastic infectious pathways [32,51].
Hofbauer cells (HCs) are human placental macrophages, which are innate immune cells that may penetrate trophoblasts. HCs may also produce IFN-α and proinflammatory cytokines and activate antiviral gene expression but do not cause cell death [32,52]. However, during an in vitro study, cells from a mature placenta were isolated, and it was found that HCs and cytotrophoblasts could be accessed by ZIKV. After ZIKV infection, cytotrophoblasts and macrophages promote an increase in viral transcript levels, which may lead to fetal pathogenesis [39,52]. Conversely, as the pregnancy progresses, the placenta undergoes morphological changes, reducing the cytotrophoblast layer, hence, becoming less susceptible to ZIKV infection. Therefore, it is notable that during ZIKV infection, the changes in fetal brain formation occurs primarily in the first trimester of pregnancy [32].
ZIKV and the immune system
Innate immune responses are regulated by protein tyrosine kinases (PTK), such as TAM receptors. TAM plays an important role in the elimination of apoptotic cells and natural killer (NK) cell differentiation. Furthermore, AXL and MER receptors are recognized by macrophages and monocytes, antigen-presenting cells [43,53]. These receptors are related to the inhibition of the innate immune response during ZIKV infection [43,54-56]. For successful ZIKV infection, Gas6 (growth-arrest-specific 6) and S protein must bind to mediate virus entry into the cell (Figure 2). Following the formation of the virus–Gas6/ProS complex, recruitment of the interferon receptor (IFNAR) causes the expression of SOCS 1 and SOCS 3 (suppressor of cytokine signaling) and, consequently, inhibition of inflammatory cytokines, thus, inhibiting the system and facilitating viral replication [43].
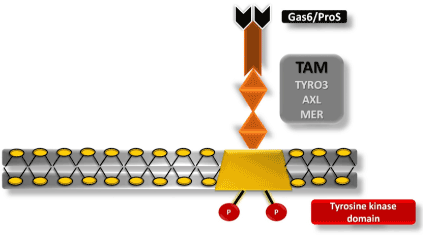
Figure 2. Tyrosine kinase receptor, TAM. Schematic representation of the cell membrane of a CNS cell containing a TAM receptor (TYRO3, AXL, and MER) interacting with a Gas6/ProS ligand necessary for the interaction of ZIKV with the host cell, inhibition of the immune system, and subsequent infection.
Molecular interaction of ZIKV with fetal CNS cells
Fetal CNS infection caused by ZIKV occurs through the rupture of the placental barrier and entry into the developing brain through hematogenous dissemination or cerebrospinal fluid [57,58]. Considering that the AXL receptor is expressed in cells of the radial glia, microglia, and astrocytes, which are present in the developing human cortex [58,59]. Evidence indicates that ZIKV preferentially enters via this group of cells, particularly fetal radial glial cells. While multiple possible viral entry receptor mechanisms have been proposed, several lines of evidence support the AXL receptor as playing a particularly important role [41,42]. Radial glial cells are considered as primary neural stem cells in the human brain; they span the entire developing cortex and are connected to the vascular and cerebrospinal fluid compartments. AXL signaling is usually able to maintain neurogenesis, survival, and proliferation of neural stem cells. In addition, this receptor maintains the blood–brain barrier to protect the host against the neurotropism of some viruses [58,60]. The AXL receptor can be detected at a high level during the first 20 weeks of gestation, particularly in the microglia, ventricular zone (VZ), and subventricular zone (SVZ). As gestation progresses, these microglial cells became more condensed in the VZ and SVZ regions [58,59,61]. Infection of the neural progenitor cells (NPCs) leads to the deregulation of the cell cycle and subsequent cell death, resulting in microcephaly and other changes in the CNS [48]. Thus, when ZIKV binds to AXL via the Gas6 ligand, it resembles the regular activation and signaling of AXL (Figure 3). This process inhibits the innate immune response and enables more effective viral infection [58,59,62].
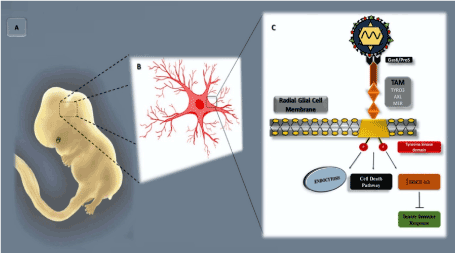
Figure 3. ZIKV–fetus molecular interaction. A Representation of a human fetus at approximately 8 weeks of development, highlighting the CNS developmental region. B Schematic representation of the radial glia cell, with prominent focus on the region of the plasma membrane. C Schematic representation of the part of the plasma membrane of the radial glial cell, presenting a tyrosine kinase (TK) receptor with its respective TYRO3, AXL, and MER (TAM) receptors and a Gas6/ProS ligand (in black) interacting with the viral envelope membrane protein, phosphatidyl serine (PtdSer) (in red)
Despite an increasing number of studies describing the outcomes of ZIKV infection, further detailed studies need to be conducted to assess the infection mechanism, particularly in the long-term, in infected children [63]. The effort to prevent the viral ZIKV infection process and/or its virulence factors haves been made on several fronts. For example, it is known that several host cellular pathways, such as AMP-activated protein kinase (AMPK) and mitogen-activated protein kinase (MAPK), are exploited by flaviviruses for their proper replication or proliferation [64]. Recently, it has been reported that the protein kinase A (PKA) pathway is, somehow, involved in the ZIKV replication process. The authors stated that a PKA inhibitor (PKI) is effective in suppressing ZIKV replication with minimal cytotoxicity [65]. ZIKV infection is also related to the innate immune system by the TLR3 pathway activation in in vitro human embryonic stem cell-derived cerebral organoids and, consequently, pro-apoptotic pathway activation. Interestingly, TLR3 inhibition reduces the phenotypic effects of ZIKV infection in cerebral organoids [66]. Neuronal death and other symptoms associated with ZIKV are also prevented by blockade blocking of the N-methyl-D-aspartate receptor (NMDAR) by using memantine and other NMDAR inhibitors [67,68]. Furthermore, several drugs and possible treatments have been developed and tested with different strategies to avoid virus infection and, consequently, maternal–fetal transmission, as described in the review by Gorshkov et al. [69]. Regarding the interaction between ZIKV and cellular membrane receptors, studies have indicated that inhibitors acting on AXL may be protective against ZIKV infection. Inhibitors, such as MYD1 and R428, display antiviral activity. MYD1 blocks ZIKV interaction with AXL to prevent Gas6 interaction, thus, preventing AXL signaling. In contrast, the R428 kinase inhibitor blocks AXL phosphorylation, which increases signaling of the innate immune response after infection. In addition, neutralizing antibodies or small interfering RNAs (RNAi) targeting the AXL receptor and/or expression thereof dramatically reduces ZIKV infection in primary dermal fibroblasts. However, the effective role of the AXL receptor in ZIKV infection of nerve cells is yet to be totally determined [41]. Studies have suggested that ZIKV may have a range of different possible receptors, and that AXL is more related with the suppression of the innate immune system by the inhibition of type I IFN cytokine signaling than with cell entry itself [70]. Furthermore, several recent studies, by using different biological models, have shown that AXL is not the key receptor for ZIKV infection, but it has a major role for infection in some special cell types [55,70-73].
Despite worldwide concern about the consequences of ZIKV infection, its neural stem cell tropism, and the devastating mechanism that leads to cell death, something extremely positive can be inferred from this serious scenario. Remarkably, a recent study used a Brazilian strain of ZIKV as an alternative approach to oncolytic viral therapy [74]. Because ZIKV presents tropism of NPCs and aggressive CNS embryonal tumors share several morphological and structural characteristics with NPCs, the authors showed that ZIKV can infect and kill stem cell-like cancer cells in aggressive human embryonic tumors. In addition, this approach is quite promising as a single injection of ZIKV to BALB/c nude mice resulted in significantly longer survival, decreased tumor burden, metastasis reduction, and complete tumor remission in some animals [74].
The problems observed worldwide in relation to ZIKV infection highlights vulnerabilities in the epidemic control, particularly in underdeveloped countries. Severe neurological disorders, such as GBS, encephalitis, myelitis, stillbirths, and newborns with microcephaly, in addition to the devastating effects on family and economics, justify a global effort to understand the process of virus infection and inhibit it. Identifying the potential interaction targets of ZIKV may be an excellent strategy for the development of effective infection inhibitors as no vaccine is available yet against ZIKV. Even after its probable development, the vaccine or infection inhibitors may not reach all communities susceptible to the virus, particularly in poor and/or underdeveloped countries. The lack of basic sanitation, inefficiency in public policies against vectors, and raising public awareness that minimizes mosquito proliferation and, hence, virus spread, is still a great challenge to minimize the effects caused by ZIKV infection.
This study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Director Board and Research department of Morgana Potrich College.
- Schatzmayr HG (2001) Emerging and re-emerging viral diseases. Cad Saude Publica 17: 209-213.
- Diniz D, Brito L (2016) Zika virus disease epidemic: Information and knowledge TT. Rev Eletronica Comun Inf Inov 10: 1-5.
- Hayes EB (2009) Zika virus outside Africa. Emerg Infect Dis 15: 1347-1350. [Crossref]
- Menezes R, Bandeira RM (2016) Integrated biology and english activity on the themezika virus and microcephaly. National congress of education
- Musso D, Nilles EJ, Cao-Lormeau VM (2014) Rapid spread of emerging Zika virus in the Pacific area. Clin Microbiol Infect 20: O595-596. [Crossref]
- Schuler faccini L, Ribeiro EM, Feitosa IML, Horovitz DDG, Cavalcanti DP (2016) Possible association between zika virus infection and microcephaly brazil. Morb Mortal Wkly Rep 65: 59-62.
- Campos JM, Oliveira DM, Neto C (2018) Arboviruses of epidemiological importance in Brazil. Rev Basic Health Sciences and Apl 1: 36-48.
- Vasconcelos PF (2003) Yellow Fever. Rev Soc Bras Med Trop 36: 275-293. [Crossref]
- Atif M, Azeem M, Sarwar MR, Bashir A (2016) Zika virus disease: a current review of the literature. Infection 44: 695-705.
- Aziz H (2017) Zika virus: Global health challenge, threat and current situation. J Med Virol 89: 943-951.
- Ribeiro BNF, Muniz BC, Gasparetto EL, Ventura N, Marchiori E (2017) Congenital zika syndrome and neuroimaging findings: what do we know so far?. Radiol Bras 50: 314-322. [Crossref]
- Yadav S, Rawal G, Baxi M (2016) Zika Virus: An Emergence of a New Arbovirus. J Clin Diagn Res 10: 01-03. [Crossref]
- de Oliveira CS, da Costa Vasconcelos PF (2016) Microcephaly and zika virus. J Pediatr (Rio J) 92: 103-105. [Crossref]
- Munjal A (2017) Advances in developing therapies to combat zika virus: Current knowledge and future perspectives. Front Microbiol 8: 1469.
- Macnamara FN (1954) Zika Virus: A report on three cases of human infection during an epidemic of Jaundice in Nigeria. Trans R Soc Trop Med Hyg 48: 139-145.
- Abushouk AI, Negida A, Ahmed H (2016) An updated review of Zika virus. J Clin Virol 84: 53-58. [Crossref]
- Basundra S, Hiremath RN, Khajuria R, Ghodke S (2016) Zika virus: An emerging public health challenge. J Krishna Inst Med Sci Univ 5: 5-12.
- Blazquez AB, Sainz JC (2016) Neurological manifestations of congenital Zika virus infection. World J Virol 5: 135-143.
- Carneiro RG (2017) Zika, a research agenda for (thinking) in social and human sciences in health. Interface Commun Heal Educ 21: 753-757.
- Cao-lormeau VM (2016) Guillain-Barre syndrome outbreak caused by zika virus infection in french polynesia. Lancet 387: 1531-1539.
- Araujo AQC, Silva MTT, Araujo APQC (2016) Zika virus-associated neurological disorders: A review. Brain 139: 2122-2130.
- Ministerio da Saude (2015) Epidemiological situation of the occurrence of microcephaly in Brazil. Bol. Epidemiologico 46: 1-3.
- McNeil CJ, Shetty AK (2017) Zika Virus: A Serious Global Health Threat. J Trop Pediatr 63: 242-248. [Crossref]
- Ministério da Saude (2017) Zika virus in Brazil. The SUS response. Ministry of Health Health Surveillance Secretariat.
- Coes M (2015) Monitoring of microcephaly cases in brazil. Ministry of Health. Secr Health Surveillance 4: 1-11.
- European Centre for Disease Prevention and Control (2015) Rapid risk assessment: Zika virus epidemic in the Americas: potential association with microcephaly and Guillain-Barre syndrome.
- Albuquerque MFPM, Souza WV, Araújo TVB, Braga MC, Miranda Filho DB, et al. (2018) The microcephaly epidemic and Zika virus: building knowledge in epidemiology. Cad Saude Publica 34: e00069018. [Crossref]
- Cauchemez S, Besnard M, Bompard P, Dub T, Guillemette-Artur P, et al. (2016) Association between Zika virus and microcephaly in french polynesia, 2013-15: A retrospective study. Lancet 387: 2125-2132. [Crossref]
- Barrows NJ, Campos RK, Liao KC (2018) Biochemistry and molecular biology of flaviviruses. Chem Rev 118: 4448-4482. [Crossref]
- Lindenbach BD, Rice CM (2003) Molecular biology of flaviviruses. Advances in Viruses Research 1: 1-10.
- Singh RK (2016) Zika virus - emergence, evolution, pathology, diagnosis, and control: current global scenario and future perspectives - a comprehensive review. Vet Q 36: 150-175.
- Olagnier D, Muscolini M, Coyne CB, Diamond MS (2016) Mechanisms of Zika Virus Infection and Neuropathogenesis. DNA Cell Biol 35: 367-372. [Crossref]
- Da Silva E (2017) Zika Virus: Evolutionary factors determining its epidemic and pathogenesis. Rev Saude Integr 19: 51-59.
- Faye O, Freire CC, Iamarino A, Faye O, de Oliveira JV, et al. (2014) Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl Trop Dis 8: e2636. [Crossref]
- Haddow AD (2012) Genetic characterization of zika virus strains: Geographic expansion of the asian lineage. PLoS Negl Trop Dis 6: 1-10.
- Sirohi D (2016) The 3.8 A resolution cryo-EM structure of zika virus. Science 80: 1-10.
- Beasley DWC (2005) Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J Virol 79: 8339-8347.
- Giovanetti M (2016) Zika Virus spreading in south america: Evolutionary analysis of emerging neutralizing resistant Phe279Ser strains. Asian Pac J Trop Med 9: 445-452.
- Rabelo K (2017) Outcomes of zika virus infection outbreaks. Rev Saude Fisica Ment 5: 61-75.
- Xie X, Gayen S, Kang C, Yuan Z, Shi PY (2013) Membrane topology and function of dengue virus NS2A protein. J Virol 87: 4609-4622. [Crossref]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, et al. (2015) Biology of zika virus infection in human skin cells. J Virol 89: 8880-8896. [Crossref]
- Meertens L (2012) The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12: 544-557.
- Perera-Lecoin M, Meertens L, Carnec X, Amara A (2014) Flavivirus entry receptors: An update. Viruses 6: 69-88.
- Tabata T (2016) Zika virus targets different primary human placental cells suggesting two routes for vertical transmission. Cell Host Microbe 20: 1-28.
- Shafit-Zagardo B, Gruber RC, DuBois JC (2018) The role of TAM family receptors and ligands in the nervous system: From development to pathobiology. Pharmacol Ther 188: 97-117.
- Calvet G, Aguiar RS, Melo ASO, Sampaio SA, de Filippis I, et al. (2016) Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study. Lancet Infect Dis 16: 653-660. [Crossref]
- Driggers RW (2016) Zika virus infection with prolonged maternal viremia and fetal brain abnormalities. N Engl J Med 374: 2142-2151.
- Miner JJ, Diamond MS (2016) Understanding how zika virus enters and infects neural target cells. Cell Stem Cell 18: 559-560.
- de Noronha L, Zanluca C, Azevedo MLV, Luz KG, dos Santos CND (2016) Zika virus damages the human placental barrier and presents marked fetal neurotropism. Mem Inst Oswaldo Cruz 111: 287-293.
- Bayer A, Lennemann NJ (2016) Type III interferons produced by human placental trophoblasts confer protection against zika virus infection. Cell Host Microbe 19: 1-17.
- Delorme-Axford E (2013) Human placental trophoblasts confer viral resistance to recipient cells. Proc Natl Acad Sci 110: 12048-12053.
- Quicke KM (2016) Zika virus infects human placental macrophages. Cell Host Microbe 5: 1-10.
- Rothlin CV, Lemke G (2010) TAM receptor signaling and autoimmune disease. Current Opinion in Immunology 1: 1-10.
- Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, et al. (2003) Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol 4: 87-91. [Crossref]
- Hastings AK, Yockey LJ, Jagger BW, Hwang J, Uraki R, et al. (2017) TAM receptors are not required for zika virus infection in mice. Cell Rep 19: 558-568. [Crossref]
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MBA, Lemke G (2007) TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131: 1124-1136.
- Brasil P, Pereira JP, Moreira ME, Ribeiro Nogueira RM, Damasceno L, et al. (2016) Zika virus infection in pregnant women in Rio de Janeiro. N Engl J Med 375: 2321-2334. [Crossref]
- Nowakowski TJ (2016) Expression analysis highlights AXL as a candidate zika virus entry receptor in neural stem cells. Cell Stem Cell 18: 591-596.
- Meertens L (2017) Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep 18: 324-333.
- Miner JJ (2015) The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat Med 21: 1464-1472.
- Ji R (2013) TAM receptors affect adult brain neurogenesis by negative regulation of microglial cell activation. J Immunol 191: 6165-6177.
- Mlakar J (2016) Zika Virus Associated with Microcephaly. N Engl J Med 374: 951-958. [Crossref]
- Lebov JF (2018) Review: Evidence of neurological sequelae in children with acquired zika virus infection. Pediatr. Neurol 85: 16-20.
- Airo AM (2018) Expression of flavivirus capsids enhance the cellular environment for viral replication by activating Akt-signalling pathways. Virology 516: 147-157.
- Fan Cheng, Suzane Ramos da Silva, Chueh Huang, Jae U Jung, Shou Jiang Gao (2018) Suppression of zika virus infection and replication. Jounal Virol 92: 1-17.
- Dang J (2016) Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 19: 258-265.
- Costa VV (2017) N-Methyl-D-Aspartate (NMDA) receptor blockade prevents neuronal death induced by zika virus infection. Am Soc Microbiol 8: 1-16
- Gaburro J (2018) Zika virus-induced hyper excitation precedes death of mouse primary neuron. Virol J 15: 1-10.
- Gorshkov K (2019) Zika virus: Origins, pathological action, and treatment strategies. Front Microbiol 10: 1-17.
- Chen J, Yang YF, Yang Y, Zou P, Chen J, et al. (2018) AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat Microbiol 3: 302-309. [Crossref]
- Wang ZY, Yang WL, Li DJ, Chen W, Zhao Q, et al. (2019) Comparison of biometry with the Pentacam AXL, IOLMaster 700 and IOLMaster 500 in cataract patients. Zhonghua Yan Ke Za Zhi 55: 515-521. [Crossref]
- Wells MF (2016) Genetic ablation of axl does not protect human neural progenitor cells and cerebral organoids from zika virus infection. Cell Stem Cell 19: 703-708.
- Wang ZY, Wang Z, Zhen ZD, Feng KH, Guo J, et al. (2017) Axl is not an indispensable factor for Zika virus infection in mice. J Gen Virol 98: 2061-2068. [Crossref]
- Kaid C, Goulart E, Caires-Júnior LC, Araujo BHS, Soares-Schanoski A, et al. (2018) Zika virus selectively kills aggressive human embryonal cns tumor cells in vitro and in vivo. Cancer Res 78: 3363-3374. [Crossref]