Abstract
The acute-phase protein α1-antirypsin (AAT) has recently been suggested to exert beneficial effects in various immune-associated pathologies, such as graft-versus-host disease, rheumatoid arthritis, multiple sclerosis and allogeneic transplantation. These effects have been demonstrated to take place through direct immunomodulation of dendritic cells, macrophages, neutrophils and B cells, while allowing, in a yet inexplicable distinction, intact isolated functions of T cells and NK cells. With such a unique discriminatory targeting of immunocytes, AAT drives immune responses simultaneously towards regulation of inflammation and expansion of antigen-specific regulatory T cells (Tregs). Based on this intriguing activity profile, a concern was raised regarding the impact of chronic treatment with human AAT with respect to susceptibility to tumors and to metastatic spread. Tumor development is linked to inflammatory responses in a manner which largely promotes immune evasion. Local innate immunocytes, such as tumor-associated macrophages (TAMs), tumor-associated dendritic cells (TADCs) and the recently appreciated tumor-associated neutrophils (TANs), are considered instrumental in primary tumor survival. This is accomplished in part by the release of growth factors and cytokines, including VEGF, TGFβ, IL-10 and IL-1 receptor antagonist (IL-1Ra), all of which have been demonstrated to be elevated in inflammatory conditions during AAT therapy. While clinical studies report heightened serum AAT levels in patients with a variety of advanced tumors, there is no evidence to point to a particular pro-tumor effect of AAT, and the possibility that its elevation in the blood might represent a meresystemic marker– rather than an accessory to tumor development– is grossly overlooked. Indeed, preclinical studies reveal significant inhibition of tumor development during treatment with AAT, and prolonged follow-up studies of individuals who receive life-long excessive doses of intravenous AAT do not depict a rise in tumor occurrence. Considering the prospect of AAT being indicated for medical indications to individuals with normal levels of the protein, its relation to tumor immunology is of immense importance. We hereby review the current knowledge regarding some possible roles that AAT may play, both locally and systemically, in the delicate and detrimental arena of tumor immunology.
Key words
inflammation, cancer, immunomodulation
Introduction
Alpha1-antitrypsin (AAT) is a 52 kDa anti-inflammatory glycoprotein that has recently come to focus as an immune modulator [1,2]. Clinically relevant settings for harnessing its benefits include recent onset autoimmune diabetes [1], immunosuppression-resistant graft-versus-host disease (GvHD) [3,4], allograft transplant rejection [5], multiple sclerosis [6] and inflammatory bowel diseases [7]. At present, over half a dozen clinical trials evaluate the outcomes of infusing clinical-grade AAT to individuals with no genetic deficiency in AAT, in the context of modulating unwanted immune responses[1].
The activity most identified with AAT is inhibition of inflammation-facilitating serine-proteases, such as neutrophil elastase, cathepsin G and proteinase-3 (PR3) [8]. AAT is furthermore capable of inhibiting the activity of certain non-serine proteases, including caspase-3 in pancreatic β cells [9]. Another non-serine protease target of AAT includes MMP-9 both in a direct manner and an indirect one; MMP activation requires cleavage of pro-MMP by the main substrates of AAT [1], and also, AAT is cleaved and inactivated by members of the MMP family (MMP-1,3,7,8,11) [10,11].
A common circulating protein, serum AAT levels rise four- to six-fold during acute phase responses [1]. Indeed, the promoter of the gene for AAT is responsive to the IL-1 pathway, as well as to the IL-6 pathway and to hypoxia. The source of circulating AAT is primarily liver cells, yet AAT is also synthesized by lung and colon cells, presumably augmenting its presence in mucosal tissues. AAT may also be expressed by macrophages under particular conditions [12]. Interestingly, inflammatory oxidative bursts neutralize the anti-proteolytic function of AAT, allowing activated neutrophils and macrophages to migrate and to exert bacteriocidic functions [13]. Indeed, in the past thirty years, life-long weekly infusions of plasma-derived affinity-purified AAT have become the treatment of choice for AAT deficient individuals [14] and, despite prolonged exposure to excessive circulating AAT and its renowned anti-inflammatory profile, long-term studies indicate that treated patients exhibit an unexpected reduction in bacterial infection rates [1,15].
Various studies, including several conducted by our group, have established that AAT exerts not only anti-inflammatory activities, but also antigen-specific immunoregulatory activities, as evident upon examining the collective outcomes of AAT treatment in several major in vivo models and immune cell types. For example, B lymphocytes [16], macrophages [17] and dendritic cells (DCs) [18] appear to become less inflammatory and more tolerogenic under treatment with AAT; of these, DCs display reduced expression and surface presentation of IL-15 [19] while they upregulate the secretion of IL-10 and their migratory receptor CCR7 [18], and B lymphocytes exhibit diminished proliferation and compromised isotype switching while maintaining potent IgM secretion [16]. In contrast, isolated T cells and NK cells appear to be unaffected by AAT, thus NK cells and regulatory T cells (Tregs) are presumed to be modulated indirectly by virtue of the tolerogenic effects of AAT on DCs and macrophages [18,19]. Indeed, the immune tolerance induced by AAT in models of allogeneic islet transplantation is suggested to be mediated by alloantigen-recognizing DC-activated Tregs, which maintain long-term antigen-specific tolerance towards grafted allogeneic cells [20].
Cancer and circulating AAT: is there an apparent association?
Individuals that carry a genetic deficiency in AAT are at risk of developing lung emphysema, suggesting that an added role for AAT may exist, that is, the promotion of tissue repair and wound healing. Left untreated, these patients are also at risk of developing various forms of vasculitis, agreeing with the major biological interface in which AAT functions, i.e., the endothelial lining. However the deficiency in levels of circulating AAT are not the primary mechanism of pathology in these cases; the most damaging form of AAT deficiency, the Z allele, results in intracellular aggregates of improperly folded AAT in both liver and lung cells, concomitant with reduced serum AAT levels[21]. Intriguingly, AAT deficient individuals are also at risk for developing tumors of the liver [22,23], lung [24,25] and gastrointestinal (GI) tract [26]. Considering these aforementioned cellular aspect in AAT deficiency, these pathologies may result both from intracellular damage endured by AAT-producing cells, as well as by inappropriate low levels of circulating AAT [27]; whether replacement therapy with AAT diminishes these collective risks is yet to be determined.
A mechanistic role for AAT in cancer biology and immunology is strikingly lacking in the literature, and the presently available studies might be misleading. On the one hand, a rise in serum levels of AAT and other acute-phase proteins has been documented in various forms and stages of cancer, including hepatocellular carcinoma [28], lung cancer [29,30], GI tract tumors [31], breast cancer [32], pancreatic cancer [33] and urinary bladder cancer [34]; on the other hand, these might represent the inflammatory response of a disease-stricken body. Similarly, in some cases, a positive correlation between serum or local AAT levels and tumor progression can be documented, alongside a negative prognosis. Upon deeper examination, certain superficial molecular modifications in AAT have also been reported to occur in cancer patients, including excessive glycosylation [35] and increased fucosylation in patients with ovarian cancer [36], hepatocellular carcinoma (HCC) and liver cirrhosis [37]. These studies, without exception, have regarded circulating AAT as a molecularmarker of cancer associated-inflammation, rather than a functional molecule.
Certain in vitro studies provide observations of a more mechanistic value for AAT: AAT was shown to inhibit tumor growth and tumor-elicited angiogenesis in nude mice [38], and the C-terminal portion of AAT that is released upon proteolytic cleavage (C36) inhibits NK cell activity in vitro; consistently, a C36–expressing pancreatic adenocarcinoma cell line proved more invasive, in vivo [10], yet there is no evidence to support the presence of native C36 in in vivo tumor models. In contrast, a study which employed a model in which a HCC cell line was treated with AAT and C36 in conjugation with neutrophil-conditioned medium, reported significant reduction in tumor invasiveness, [39]. Such an outcome is highly unexpected when considering that AAT caused an increase in VEGF expression, as corroborated in angiogenesis-related studies. In line with these findings, a series of studies centered on the MCF-7 human breast cancer cell line and revealed a negative correlation between AAT production by tumor cells and tumor cell proliferation; this was accompanied by a reduction in active TGFβ release and in in vivo invasiveness [40-42].
Circulating AAT, in the context of cancer, may thus introduce important aspects that relate to the various newly-appreciated functions of AAT. At least one of such activities has to do with the apparent association between AAT and TGFβ activities. TGFβ is expressed by a multitude of cellular populations, including M2-like macrophages and Tregs [43], and regulates differentiation, development and homeostasis in nearly all types of tissues [44]. TGFβ expression positively correlates with late stage tumor promotion, and the involvement of TGFβ in tumor progression and recurrence by way of promotion of immune evasion has been well documented [44]. For example, TGFβ interferes with naïve T cell differentiation into mature CD4+ T helper cells and CD8+ cytotoxic T cells [45] and induces the differentiation of tumor-promoting CD4+ Foxp3+ Tregs [45]. TGFβ signaling is a highly regulated process which involves proteolytic cleavage of the inactive TGFβ proprotein; this cleavage is conducted by several families of proteases, which include MMPs (such as MMP-9 and MMP-2) [46], cathepsins [46] and proteinase 3 (PR3) [47]. Evidence points to effective inhibition of these proteases by AAT [1]. Nonetheless, when considering the influence of AAT on TGFβ, one must consider the involvement of several layers of AAT-related regulation. While AAT may directly inhibit proteases responsible for cleavage and activation of mature TGFβ, AAT may also indirectly increase local TGFβ levels by promoting the polarization of macrophages towards M2 profile. Indeed, while in vitro treatment of isolated monocultures of human breast cancer cells MCF-7 with AAT resulted in lower levels of mature TGFβ [40,41], in vivo studies demonstrate that AAT treatment results in an elevation of TGFβ levels [48], correlating with a shift in macrophage polarization towards M2 profile [20]. An example for the important of context in interpreting the activities of AAT involves an in vivo model of liver fibrosis, where hepatic synthesis of TGFβ is found to be significantly higher in transgenic mice carrying the Z allele of AAT [49]; in order to fully interpret this outcome one should take into consideration that the model incorporates acute liver inflammation, as instigated by intracellular aggregates of AAT, rather than a possible effect of reduced circulating levels of AAT.
AAT inhibits IL-6 production [50,51]. An instigator of systemic inflammation, as well as a major pleiotropic agent, IL-6 is also involved in neoplastic progression [52] and immune evasion [53,54], and may assist tumor cells in overcoming chemotherapy [52,53]. Considerable efforts have been made to block IL-6 activity in the context of tumor therapy [55-58]; thus, the fact that AAT downplays IL-6 levels represents one of several possible mechanisms of action in favor of interfering with tumor growth. However, reduced inflammation might also present as a window of opportunity for immune evasion by newly formed cancerous foci, and the conditions in favor of Treg expansion (i.e., IL-1¯, IL-6¯, TGFb) might also preclude an effective immune response against cancer cells. Yet, treatment with excessive doses of AAT does not expose patients to cancer.
AAT is a highly selective immunomodulator that allows T cell and NK cell responses
All studies that examined IL-2–driven isolated T cell responses have reported intact T cell activation in the presence of AAT, separating AAT categorically from classic immunosuppression [20]. An intact T cell and NK cell population is undoubtedly a positive factor in eradicating tumors; yet it is an unexpected observation that neither cell types, once in an isolated or enriched culture, alter their response in the presence of AAT. As in T cells, NK cell activation is an intricate process,the sum of activating and inhibitory membranal signals. Among the best characterized NK cell activating receptors are NKG2D and NKp46. NKG2D is expressed by NK cells as well as αβ and γδ T cells, and is considered central to recognition of ligands expressed by cells that have undergone malignant transformation or viral infection [59]. NKp46, on the other hand, is expressed solely by NK cells, and is critical for NK cell recognition of viral haemagglutinins associated with infection by influenza virus, as well as recognition of tumor cells, as exemplified by effective inhibition of NK cell cytotoxicity by NKp46-blocking monoclonal antibodies [60]. Importantly, NKp46 was recently proven to be the NK cell receptor responsible for recognition and subsequent cytotoxicity against pancreatic β cells in the context of autoimmune type 1 diabetes [61-63].
Expression of the activating receptors NKp46 and NKG2D by NK cells has recently been studied. According to the report [19], both receptors are unaffected by short-term in vivo AAT treatment, yet NK cell degranulation towards pancreatic β cells is downregulated. Interestingly, degranulation was not diminished towards AAT-treated tumor cell lines). In another study that examined a model of B cell lymphoma, AAT treatment was reported to maintain and in some respect to even enhance graft versus leukemia (GVL) response upon bone marrow transplantation [3]. This reported effect was suggested to be dependent onan expanded population of NKG2D+ NK cells and the maintenance of a CD11c+ DC population [3]; all the while, CD8+ T cells were unaffected by the presence of AAT.
CD8+ T cells are critical players in the eradication of tumor cells, as they provide both direct killing and elevate local IFNγ levels thus promoting tumor-suppressing Th1 responses [64-69]. That a native molecule holds the capacity to distinguish cytotoxic-worthy targets (such as tumor cells) from protection-worthy antigenic targets (such as allografts) is indeed perplexing. The choice of any organism to elevate systemic AAT levels during infections may, in this regard, facilitate the minimizing of self-recognition and diminish the development of aberrant cells and tumors. In addition, although IL-2–activated T cells are unaffected by AAT, the process of antigen presentation and activation of T cells by antigen-presenting cells is undoubtedly modified by AAT treatment, as activated B cells, DCs and macrophages all shift towards an IL-10–producing phenotype in its presence. While these changes may collectively explain some of the outcomes of AAT treatment in tumor models, a complete mechanism of action is still to be identified.
Local considerations: AAT modifies local antigen-presenting cell profiles
Tumor development occurs concomitantly with subversion of anti-tumor immune responses [70]. Yet a tumor mass is not absent of immune cells; tumor-associated macrophages (TAM)and tumor-associated dendritic cells (TADC) (see illustration) have been extensively studied and shown to mediate tumor progression and immune evasion in a variety of mechanisms [71-73]. Both of these cellular populations, TAMs and TADCs, are thought to originate from monocytes recruited into the tumor area from the bone marrow and to a smaller extent from the spleen. The influence of AAT on the behavior of these central players in tumor immunology has yet to be directly examined, yet various aspects of the effect of AAT on macrophages and DCs may shed light on possible ramifications of AAT treatment in the context of a tumor.
Macrophage M1 and M2 phenotypes are opposite extremities of a range of possible activation states, presumably forming a continuum rather than a binary activation pattern. While M1-activated macrophages (mainly induced by IFNγ or LPS) are a rather homogenous population that facilitates a Th1-type cyototoxic response and tumor cell killing (by secreting IL-12, IL-18 and IFNα/β). M2-activation, in contrast, is highly heterogeneous and may acquire one of several "M2-like" phenotypes, such as M2a (induced by IL-4 and IL-13), M2b (induced by IL-1β and antibody immune complexes) and M2c (induced by IL-10, TGFβ and glucocorticoids, which have long been established as inducers of AAT expression). The M2-like phenotypes are differentiated both by expression of membranal markers as well as by cytokines and trophic factors which they secrete; while M2a macrophages are associated with tissue remodeling and late-stage wound healing by way of growth factor secretion, M2c macrophages actively regulate T cell cytotoxicity and promote regulatory T cell differentiation by secreting IL-10 and TGFβ [74].
TAMs facilitate immune evasion, as well as angiogenesis and tissue remodeling by secreting growth factors and angiogenic agents, including IL-6, TGFβ and VEGF [75-77], and also immune-regulatory molecules, such as IL-10, IL-1 receptor antagonist (IL-1Ra) and the IL-1–neutralizing soluble IL-1R [43,71,78]. TAMs share numerous properties with M2-activated macrophages, but nonetheless must be viewed as a separate population to M2-activated macrophages. TAMs are considered to originally acquire M1-like properties upon infiltration to a tumor, and are then skewed towards an M2-like profile in advanced tumors as a result of exposure to factors secreted by tumor cells [43,71] or tumor-associated fibroblasts (TAF) [79], including TGFβ and IL-10. Two major hallmark attributes of macrophages are their longevity and plasticity; indeed, numerous factors are known to significantly skew macrophage activation profiles across a wide range of phenotypes [80,81]. Thus, many therapeutic approaches are based on the prospect of shifting tumor-associated macrophages towards M1 profile, rather than depleting macrophages altogether [80,81]; this is based upon the trend of minimizing systemic immunosuppression, and utilizing the highly advantageous prospect of macrophage-assisted T cell killing of tumor cells. Thus, any possibility of significantly re-directing TAM activation, e.g., by way of AAT treatment, must be considered.
Quite unexpectedly, it was recently found that AAT is expressed by macrophages under a hypoxia-activated promoter [12,82]. Its release from macrophage cells was most apparent in M1-like macrophages [83]. Added to cultures and tested in animal models, AAT is further suggested to drive macrophages towards an M2-like profile, as indicated by increased efferocytosis [84], increased IL-1Ra secretion [85] and reduced TNFα production [17]. Each immunological setting must, however, be appreciated in its larger context, especially in light of the highly differential conditions which exist in any experimental tumor model. Thus, being that hypoxic conditions lead to stabilization of hypoxia-inducible factor-1α (HIF-1α) and HIF-2α [86] and that AAT is induced by hypoxia, one must take into account the tight association between local conditions and AAT expression in the detrimental junction of TAM profile acquisition.
Much like macrophages, activated mature DCs (mDCs) are highly immunogenic cells, which specialize in antigen presentation and activation of, among others, cytotoxic T cells and NK cells, by way of IL-12, IL-18 and IFNα/β secretion and by trans-presentation of surface IL-15 [86]. Alternatively activated DCs (termed semi-mature DCs or smDCs) actively induce antigen-specific tolerance by way of either T cell deletion, energy or the promotion of Treg maturation and expansion [72,86,87]. Much like in the case of M1 and M2 macrophages, mDCs and smDCs are two opposite extremities and a continuum of possible activation states, rather than a binary programming pattern, is predicted to best reflect the state of DC maturation; indeed, numerous "intermediate" activation states have been described. In this regard, AAT has been shown to significantly favor the occurrence of smDCs [18] in allogeneic transplantation models, and thus to induce Treg maturation and transplant-specific tolerance. AAT was also found to reduce the expression and presentation of IL-15 by DCs, correlating with reduced activation of NK cells by DCs, a standalone activation pathway distinct from the apparently unaffected property of AAT-treated NK cells to directly kill tumor cells [19].
TADCs share some similarities with smDCs. Both have poor antigen-presentation capabilities and hence induce T cell anergy, Treg maturation and tumor-specific tolerance, and are therefore considered tumor-propagating cells [72,86,87]. Although TADCs share certain qualities with TAMs, they are differentiated primarily based on distribution; while TAMs are evenly distributed throughout the tumor, TADCs are primarily present in tumor periphery [72]. Indeed, deeper tumor infiltration by DCs correlates with a positive prognosis in certain tumors [88]. When considering the potential role of AAT in TADC-associated immunological responses, it is important to bear in mind that although AAT may promote DC-induced immune tolerance, the few studies that attempted to address such a pathway by AAT were conducted in models drastically different from tumor development models (i.e., allograft transplantation or autoimmune disease models). The possibility that local conditions may markedly alter the delicate relationship between AAT and myeloid cell profiles is an example of an entity not yet addressed experimentally. A detailed examination of TADC profiles and functions in AAT-treated animals is thus strongly required.
Metastatic spread as a target for inhibition by AAT
Similar to primary tumor development, metastatic spread is a process that inexorably involves inhibition and re-purposing of the immune system [89]. Although specific inhibition of metastatic spread by AAT has yet to be examined, current knowledge regarding AAT activities in separate models suggests several potential stages inherent to metastatic spread of nearly all tumor types, in which AAT may reduce the metastatic burden; each such stage will be addressed separately.
Epithelial to mesenchymal transition (EMT)
The first stage of metastatic spread is the acquisition of mesenchymal characteristics by peripheral primary tumor cells, in a process generally termed EMT [90]. Although the myriad of changes which take place during EMT are beyond the scope of this review, we note that EMT is driven by certain cytokines that originate, to a large extent, from tumor-associated leukocytes, such as TAMs and tumor associated neutrophils (TANs, see illustration) [90]. Among the best characterized cytokines which participate in EMT are TNFα and TGFβ, both of which activate the NF-kB pathway [90]. The interaction between TNFα and tumors is intricate, and may elicit both pro- and anti-tumor responses, depending upon spatial and temporal parameters. As the main source of local TNFα is derived from tumor-associated leukocytes, the association between tumor-site inflammation and TNFα-induced EMT is powerful. TNFα blockade has proven effective in blocking inflammation-associated EMT, and TNFα signaling was found to be dispensable in early-stage tumors but critical in late-stage tumors [91]. AAT has been shown to inhibit the release of membrane-associated TNFα by the ADAM17 protease, significantly reducing free TNFα levels and resulting in reduced NF-kB pathway activation [92]. Unlike TNFα, TGFβ release is more variable and can be performed by tumor cells, tumor-associated leukocytes and TAFs [90]. TGFβ was found to be associated with EMT in a variety of tumor models [90]. Nonetheless, while AAT treatment was shown to elevate TGFβ expression in models of allogeneic transplantation [20], production of TGFβ by MCF-7 cells was blocked during treatment with AAT [40-42], resulting in reduced in vivo invasiveness. As AAT may also strongly block the cleavage of pro-TGFβ, a more thorough investigation of TGFβ-associated EMT responses and their relation to AAT therapy may prove highly informative.
ECM degradation and extravasation
In order for a successful metastatic migration of tumor cells from the primary tumor to the vasculature, tumor-surrounding ECM must be degraded by proteases [93], including MMPs [94-97] cathepsins [98] and elastase [99-101], which are mainly provided by TAMs and TANs [1]. The secretion and activation of proteases by TAMs is dependent upon reprograming of TAMs [90], which may be exerted by factors secreted by tumor cells or by tumor-periphery lymphocytes, such as IL-4/13–secreting Th2 cells and IL-17–secreting Th17 cells [102,103]. In an in vitro co-culture model, breast cancer cells were observed as secreting IL-6 and inducing the production of MMP-9 and MMP-2 by monocytes [104]. All these various mechanisms share a common outcome: responding macrophages are skewed towards M2 profile characterized by a greater propensity for ECM-remodeling, which is highly conductive for tumor cell migration [90]. Direct AAT suicide-inhibition of elastase, cathepsins and certain MMPs may constitute a viable mechanism for inhibition of metastatic spread. However, AAT may also exert its influence by direct inhibition of inflammatory proteases, such as the reduction of pro-MMP cleavage. The possible influence of AAT on TAM activation may also significantly alter the local protease environment by reducing the levels of IL-6, IL-1β and TNFα in the tumor periphery.
Survival in the circulation
The process of migration and extravasation is considered a preliminary step in metastatic spread, as in most types of tumors the rate of survival of extravasated metastatic cells is low (estimated to be less than 0.01% of disseminated cells) [90]. Under certain conditions, the survival of tumor cells in the circulation is increased by inflammatory factors released from the primary tumor environment, such as IL-6 and TNFα. A proposed mechanism for this phenomenon involves tumor cell engagement with macrophages in the tumor periphery, after which the cell agglomerate becomes blood borne and tumor cells receive myeloid-induced protection from lysis by NK cells and CD8+ T cells [105]. Circulating tumor cells have also been shown to aggregate together with platelets, thus acquiring protection from NK cell recognition and lysis [106]. Platelet aggregation was found to be dependent upon expression of prothrombin and fibrinogen [106]. This mechanism correlates with reduced survivability of cancer patients with high platelet counts [107] and the reduced incidence of metastasis in animals and humans treated with anticoagulants [108]. In both mechanisms described, AAT may reduce the survivability of circulating tumor cells removed from the immune-regulatory environment of the primary tumor; lower levels of IL-6 and TNFα may reduce the propensity of TAMs to engage and extravasate with tumor cells, while the inhibition of thrombin by AAT (although inferior to antithrombin III) [109] may decrease platelet aggregation and permit intact anti-tumor NK cell responses.
Pre-metastatic niche formation
Local inflammatory conditions provide important stimuli conductive to successful metastatic spread to target organs [70,110], as they drive local macrophages (such as alveolar macrophages) to acquire a TAM phenotype [71,102,111]. Several studies have demonstrated that such inflammatory conditions may be generated by factors secreted by the primary tumor, even prior to the process of tumor cell extravasation. In a model of breast cancer lung metastasis, primary tumor-bearing mice displayed elevated populations of immature CD11b+Gr1+ myeloid cells which localized to the lungs, inhibited local IFNγ levels, increased production of IL-6, monocyte chemoattractant protein-1 (MCP-1, also termed CCL2), IL-4 and IL-10, and secreted high levels of pre-MMP9, prior to arrival of disseminated tumor cells [112]. MCP-1 in particular is considered to be a highly metastatic chemokine as it facilitates tissue monocyte infiltration, setting the ground for formation of a pre-metastatic niche [113]. In this critical context, AAT is, again, poised as a protective agent in several respects: AAT significantly inhibits monocyte activation and MCP-1 release [114], while also inhibiting MMP9 activity (Figure 1).
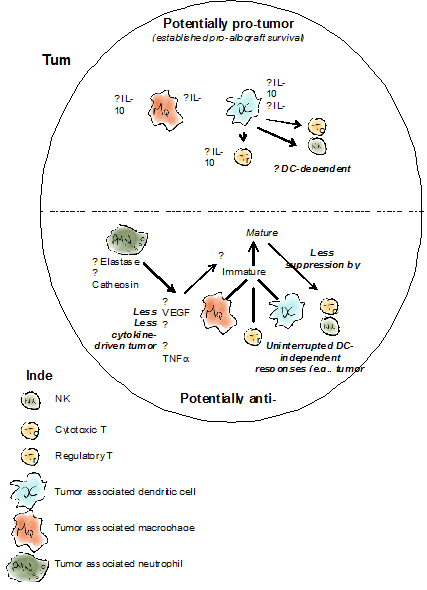
Figure 1. Suggested effects of AAT on tumor immunology based on evidence from allograft transplantation studies
Top, selected activities reported for AAT with regards to innate and adaptive immune cells and the paths that enable AAT to protect allografts. Bottom, compiled evidence from multiple reports regarding the effects of AAT on innate and adaptive immune cells in the context of advancing an anti-tumor molecular and cellular environment.
Discussion
When considering particular pathways and cellular effects exerted by AAT, one may find both pro- and anti-tumor attributes. For example, that AAT is pro-angiogenic towards islet allografts [115] is in striking contrast to its anti-angiogenic effect on tumors [38]. Similarly, being anti-inflammatory, AAT is seemingly in favor of immune evasion, as expanding Tregs and suppressing macrophage responses would appear to impose; nonetheless, when one gathers data from multiple studies and adds-up the particular paths that they address experimentally, one can observe an overall suppression of tumor growth in the presence of AAT. Indeed, the few studies that did directly examine tumor growth and AAT therapy concluded that tumors fail to thrive in its presence.
It is highly tempting to explore some clinically relevant functions of AAT by dissecting the phenotype of patients that suffer from its genetic deficiency, particularly in light of this condition being a one-gene-disease. However, it is not a one-pathway disease, and its specific pathogenesis extends beyond the mere reduced levels of circulating AAT. In a recent review by Stockley RA [116], a timely and appropriate connecting line is drawn between the clinical entity of AAT deficiency (AATD), and newly explored clinically-relevant functions of AAT infusions. Outside the immediate association between AATD and the need for augmentation therapy to restore its circulating levels, one must consider the redundancy in functions identified as AAT-related that are readily present in the circulation of AATD patients; several other serine-protease inhibitors exhibit aspects that even overlap the anti-inflammatory attributes of AAT. Examples include antithrombin III, plasminogen activator inhibitor 1, b2-macroglobulin and protein C inhibitor. Liver cell function in AATD might be affected by the pathologic intracellular content of mutated AAT, and may thus be deficient in forming a fully functional inducible cassette of acute phase response products. Therefore, aspects addressed by therapeutic introduction of AAT in the context of an aberrant immune response are not necessarily lacking in the immune system of an AATD patient. The fact that our body generates excess AAT appropriately during infection and in the third trimester of pregnancy, at levels that are largely replicated by current exogenous AAT treatment protocols, better represents the context of AAT activities in the present review. A more detailed examination of AAT activities independent of an underlying genetic deficiency, particularly in the unique circumstances of tumor immunology, is therefore very much required.
Similar to the apparent limitation in interpreting AAT activities based on its genetic deficiency, it is important to also consider that elevated levels of AAT might represent a marker, rather than an active component within the multistep pathogenesis of any given disease. While clinical studies have pointed at elevated AAT serum levels in patients with advanced cancer of several types, in all cases AAT was studied as an acute-phase protein, such that appropriately rises during systemic inflammation. Information regarding the exact effects of AAT on tumor cells and on the critical junction between innate immune cell and tumor cell interactions is quite scarce and deserves further investigation. Taken together, AAT must be examined as an independent therapeutic agent in the context of tumor immunology, without excessive reference to either its deficiency nor its elevated levels and their accompanying pathological phenotype or etiology, respectfully.
Current knowledge invariably portrays AAT as an immunomodulatory and anti-inflammatory agent. However, the models used to depict these attributes of AAT are primarily inflammatory or alloimmune responses; they do not attempt to approximate tumor-associated conditions or address tumor-specific cellular compositions. For example, one of the hallmarks of any tumor-associated environment is the chronic lack of sufficient tissue vascularization, which leads to hypoxic conditions and to the activation of hypoxia-associated pathways in macrophages and other tumor-infiltrating myeloid cell populations. Indeed, AAT expression has been shown to be increased in macrophages during hypoxic conditions in a local manner. In addition, its role in regulating tumor-associated immunology should further be divided to satisfy a categorical relation to protease inhibition; some non-protease inhibition-related activities have been described for AAT. While protease inhibition is undoubtedly an integral part of tumor-related AAT activities, e.g., tissue remodeling and metastatic extravasation, the single study that examined AAT that lacks protease inhibition capabilities found that, in vivo, tumor growth inhibition by AAT remains intact.
Taken together, collective studies in the past decade point to a potentially multifaceted immunological mechanism exerted by AAT, which should be further studied in order to elucidate the potential beneficial role of AAT in tumor immunology and to provide mechanistic evidence for consideration of AAT as a safe therapeutic for non–AAT-deficient individuals.
References
- Guttman O, Baranovski BM, Schuster R, Kaner Z, Freixo-Lima GS, et al. (2015) Acute-phase protein α1-anti-trypsin: diverting injurious innate and adaptive immune responses from non-authentic threats. Clin Exp Immunol 179: 161-172. [Crossref]
- Gottlieb PA, Alkanani AK, Michels AW, Lewis EC, Shapiro L, et al. (2014) α1-Antitrypsin therapy downregulates toll-like receptor-induced IL-1beta responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patients with type 1 diabetes. J Clin Endocrinol Metab 99: E1418-1426. [Crossref]
- Marcondes AM, Karoopongse E, Lesnikova M, Margineantu D, Welte T, et al. (2014) α-1-Antitrypsin (AAT)-modified donor cells suppress GVHD but enhance the GVL effect: a role for mitochondrial bioenergetics. Blood 124: 2881-2891. [Crossref]
- Hagen LE, Schechter T, Luk Y, Brodovitch A, Gassas A, et al. (2011) High alpha-1 antitrypsin clearance predicts severity of gut graft-versus-host disease (GVHD) in children. Pediatr Transplant 15: 659-663. [Crossref]
- Tawara I, Sun Y, Leewis EC, Toubai T, Evers R, et al. (2012) Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci U S A 109: 564-569. [Crossref]
- Subramanian S, Shahaf G, Ozeri E, Miller LM, Vandenbark AA, et al. (2011) Sustained expression of circulating human alpha-1 antitrypsin reduces inflammation, increases CD4+FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab Brain Dis 26: 107-113. [Crossref]
- Collins CB, Aherne CM, Ehrentraut SF, Gerich ME, McNamee EN, et al. (2013) Alpha-1-antitrypsin therapy ameliorates acute colitis and chronic murine ileitis. Inflamm Bowel Dis 19: 1964-1973. [Crossref]
- Breit SN, Wakefield D, Robinson JP, Luckhurst E, Clark P, et al. (1985) The role of alpha 1-antitrypsin deficiency in the pathogenesis of immune disorders. Clin Immunol Immunopathol 35: 363-380.[Crossref]
- Zhang B, Lu Y, Campbell-Thompson M, Spencer T, Wasserfall C, et al. (2007) Alpha1-antitrypsin protects beta-cells from apoptosis. Diabetes 56: 1316-1323. [Crossref]
- Kataoka H, Uchino H, Iwamura T, Seiki M, Nabeshima K, et al. (1999) Enhanced tumor growth and invasiveness in vivo by a carboxyl-terminal fragment of alpha1-proteinase inhibitor generated by matrix metalloproteinases: a possible modulatory role in natural killer cytotoxicity. Am J Pathol 154: 457-468. [Crossref]
- Okada Y, Nakanishi I (1989) Activation of matrix metalloproteinase 3 (stromelysin) and matrix metalloproteinase 2 ('gelatinase') by human neutrophil elastase and cathepsin G. FEBS Lett 249: 353-356. [Crossref]
- Carlson JA, Rogers BB, Sifers RN, Hawkins HK, Finegold MJ, et al. (1988) Multiple tissues express alpha 1-antitrypsin in transgenic mice and man. J Clin Invest 82: 26-36. [Crossref]
- Kaner Z, Ochayon DE, Shahaf G, Baranovski BM, Bahar N, et al. (2015) Acute phase protein α1-antitrypsin reduces the bacterial burden in mice by selective modulation of innate cell responses. J Infect Dis 211: 1489-1498. [Crossref]
- Brantly M, Nukiwa T, Crystal RG (1988) Molecular basis of alpha-1-antitrypsin deficiency. Am J Med 84: 13-31. [Crossref]
- Lewis EC (2012) Expanding the clinical indications for α(1)-antitrypsin therapy. Mol Med 18: 957-970. [Crossref]
- Mizrahi M, Cal P, Rosenthal M, Ochayon D, Shahaf G,et al. (2013) Human alpha1-antitrypsin modifies B-lymphocyte responses during allograft transplantation. Immunology 140: 362-373. [Crossref]
- Churg A, Wang X, Wang RD, Meixner SC, Pryzdial EL, et al. (2007) Alpha1-antitrypsin suppresses TNF-alpha and MMP-12 production by cigarette smoke-stimulated macrophages. Am J Resp Cell Mol Biol 37:144-151. [Crossref]
- Ozeri E, Mizrahi M, Shahaf G, Lewis EC (2012) α-1 antitrypsin promotes semimature, IL-10-producing and readily migrating tolerogenic dendritic cells. J Immunol 189: 146-153. [Crossref]
- Guttman O, Yossef R, Freixo-Lima G, Rider P, Porgador A, et al. (2014) α1-Antitrypsin modifies general NK cell interactions with dendritic cells and specific interactions with islet β-cells in favor of protection from autoimmune diabetes. Immunology. [Crossref]
- Lewis EC, Mizrahi M, Toledano M, DeFeliceN, Wright JL, et al. (2008) alpha1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci U S A 105: 16236-16241. [Crossref]
- Fairbanks KD, Tavill AS (2008) Liver disease in alpha 1-antitrypsin deficiency: a review. Am J Gastroenterol 103: 2136-2141. [Crossref]
- Eriksson S, Carlson J, Velez R (1986) Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N Engl J Med 314: 736-739. [Crossref]
- Rudnick DA, Perlmutter DH (2005) Alpha-1-antitrypsin deficiency: a new paradigm for hepatocellular carcinoma in genetic liver disease. Hepatology 42: 514-521. [Crossref]
- Yang P, Wentzlaff KA, Katzmann JA, Marks RS, Allen MS,et al. (1999) Alpha1-antitrypsin deficiency allele carriers among lung cancer patients. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 8: 461-465.
- Yang P, Sun Z, Krowka MJ, Aubry MC, Bamlet WR, et al. (2008) Alpha1-antitrypsin deficiency carriers, tobacco smoke, chronic obstructive pulmonary disease, and lung cancer risk. Arch Intern Med 168: 1097-1103. [Crossref]
- Yang P, Cunningham JM, Halling KC, Lesnick TG, Burgart LJ, et al. (2000) Higher risk of mismatch repair-deficient colorectal cancer in alpha(1)-antitrypsin deficiency carriers and cigarette smokers. Mol Genet Metab 71: 639-645. [Crossref]
- Sun Z, Yang P (2004) Role of imbalance between neutrophil elastase and alpha 1-antitrypsin in cancer development and progression. Lancet Oncol 5: 182-190. [Crossref]
- Sparos L, Tountas Y, Chapuis-Cellier C, Theodoropoulos G, Trichopoulos D (1984) Alpha 1-antitrypsin levels and phenotypes and hepatitis B serology in liver cancer. Br J Cancer 49: 567-570. [Crossref]
- Harris CC, Cohen MH, Connor R, Primack A, Saccomanno G, et al. (1976) Serum alpha1-antitrypsin in patients with lung cancer or abnormal sputum cytology. Cancer 38: 1655-1657. [Crossref]
- Harris CC, Primack A, Cohen MH (1974) Elevated alpha1-Antitrypsin serum levels in lung cancer patients. Cancer 34: 280-281. [Crossref]
- Karashima S, Kataoka H, Itoh H, Maruyama R, Koono M (1990) Prognostic significance of alpha-1-antitrypsin in early stage of colorectal carcinomas. Int J Cancer 45: 244-250. [Crossref]
- Lopez-Arias E, et al. (2012) Alpha 1-antitrypsin: a novel tumor-associated antigen identified in patients with early-stage breast cancer. Electrophoresis 33: 2130-2137.
- Tountas Y, Sparos L, Theodoropoulos C, Trichopoulos D (1985) Alpha 1-antitrypsin and cancer of the pancreas. Digestion 31: 37-40. [Crossref]
- Urquidi V, Goodison S, Ross S, Chang M, Dai Y, et al. (2012) Diagnostic potential of urinary alpha1-antitrypsin and apolipoprotein E in the detection of bladder cancer. J Urol 188: 2377-2383. [Crossref]
- Rostenberg I, Guízar-Vázquez J, Peñaloza R (1978) Altered carbohydrate content of alpha1-antitrypsin in patients with cancer. J Natl Cancer Inst 61: 961-965. [Crossref]
- Thompson S, Guthrie D, Turner GA (1988) Fucosylated forms of alpha-1-antitrypsin that predict unresponsiveness to chemotherapy in ovarian cancer. Br J Cancer 58: 589-593. [Crossref]
- Comunale MA, Rodemich-Betesh L, Hafner J, Wang M, Norton P, et al. (2010) Linkage specific fucosylation of alpha-1-antitrypsin in liver cirrhosis and cancer patients: implications for a biomarker of hepatocellular carcinoma. PloS One 5: e12419. [Crossref]
- Huang H, Campbell SC, Nelius T, Bedford DF, Veliceasa D, et al. (2004) Alpha1-antitrypsin inhibits angiogenesis and tumor growth. Int J Cancer 112: 1042-1048. [Crossref]
- Zelvyte I, Stevens T, Westin U, Janciauskiene S (2004) α1-antitrypsin and its C-terminal fragment attenuate effects of degranulated neutrophil-conditioned medium on lung cancer HCC cells, in vitro. Cancer Cell Int 4: 7. [Crossref]
- Finlay TH, Tamir S, Kadner SS, Cruz MR, Yavelow J, et al. (1993) alpha 1-Antitrypsin- and anchorage-independent growth of MCF-7 breast cancer cells. Endocrinology 133: 996-1002. [Crossref]
- Tamir S, Kadner SS, Katz J, Finlay TH (1990) Regulation of antitrypsin and antichymotrypsin synthesis by MCF-7 breast cancer cell sublines. Endocrinology 127: 1319-1328. [Crossref]
- Yavelow J, Tuccillo A, Kadner SS, Katz J, Finlay TH (1997) Alpha 1-antitrypsin blocks the release of transforming growth factor-alpha from MCF-7 human breast cancer cells. J Clin Endocrinol Metab 82: 745-752. [Crossref]
- Solinas G, Germano G, Mantovani A, Allavena P (2009) Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 86: 1065-1073. [Crossref]
- Principe DR, Doll J2021 Copyright OAT. All rights reserv (2014) TGF-β: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst 106: djt369. [Crossref]
- Laouar Y, Sutterwala FS, Gorelik L, Flavell RA (2005) Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol 6: 600-607. [Crossref]
- Yu Q, Stamenkovic I (2000) Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev 14: 163-176. [Crossref]
- Csernok E, Szymkowiak CH, Mistry N, Daha MR, Gross WL,et al. (1996) Transforming growth factor-beta (TGF-beta) expression and interaction with proteinase 3 (PR3) in anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Clin Exp Immunol 105: 104-111. [Crossref]
- Serban KA,Petrusca DN, Lockett AD, Justice MJ, Saint L, et al. (2015) Alpha-1 Antitrypsin (AAT) Enhances Alveolar Macrophages Scavenging During Conditions Simulating COPD Exacerbation. B110. COPD GALORE: NEW INSIGHTS INTO BRONCHITIS AND EMPHYSEMA DEVELOPMENT AND TREATMENT. American Thoracic Society International Conference Abstracts, (American Thoracic Society): A3882-A3882.
- Mencin A, Seki E, Osawa Y, Kodama Y, De Minicis S, et al. (2007) Alpha-1 antitrypsin Z protein (PiZ) increases hepatic fibrosis in a murine model of cholestasis. Hepatology 46: 1443-1452. [Crossref]
- Subramanian S, Shahaf G, Ozeri E, Miller LM, Vandenbark AA, et al. (2011) Sustained expression of circulating human alpha-1 antitrypsin reduces inflammation, increases CD4+FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab Brain Dis 26: 107-113. [Crossref]
- Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, et al. (2012) Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci U S A 109: 564-569. [Crossref]
- Hodge DR, Hurt EM, Farrar WL (2005) The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 41: 2502-2512. [Crossref]
- Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, et al. (2009) IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15: 103-113. [Crossref]
- Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454: 436-444. [Crossref]
- Grivennikov SI, Karin M (2011) Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann Rheum Dis 70: i104-108. [Crossref]
- Locasale JW, Zeskind B (2011) IL-6 and ovarian cancer--letter. Clin Cancer Res 17: 7837. [Crossref]
- Neurath MF, Finotto S (2011) IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev 22: 83-89. [Crossref]
- Petrache I, Fijalkowska I, Zhen L, Medler TR, Brown E, et al. (2006) A novel antiapoptotic role for alpha1-antitrypsin in the prevention of pulmonary emphysema. Am J Respir Crit Care Med 173: 1222-1228. [Crossref]
- Raulet DH (2003) Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 3: 781-790. [Crossref]
- Arnon TI, Achdout H, Lieberman N, Gazit R, Gonen-Gross T, et al. (2004) The mechanisms controlling the recognition of tumor- and virus-infected cells by NKp46. Blood 103: 664-672. [Crossref]
- Gur C, Enk J, Kassem SA, Suissa Y, Magenheim J, et al. (2011) Recognition and killing of human and murine pancreatic beta cells by the NK receptor NKp46. J Immunol 187: 3096-3103. [Crossref]
- Gur C, Porgador A, Elboim M, Gazit R, Mizrahi S, et al. (2010) The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol 11: 121-128. [Crossref]
- Yossef R, Gur C, Shemesh A, Guttman O, Hadad U, et al. (2015) Targeting natural killer cell reactivity by employing antibody to NKp46: implications for type 1 diabetes. PLoS One 10: e0118936. [Crossref]
- Lewis EC, Shapiro L, Bowers OJ, Dinarello CA (2005) Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci U S A 102: 12153-12158. [Crossref]
- Lu Y, Tang M, Wasserfall C, Kou Z, Campbell-Thompson M, et al. (2006) Alpha1-antitrypsin gene therapy modulates cellular immunity and efficiently prevents type 1 diabetes in nonobese diabetic mice. Hum Gene Ther 17: 625-634. [Crossref]
- Koulmanda M, Bhasin M, Hoffman L, Fan Z, Qipo A, et al. (2008) Curative and beta cell regenerative effects of alpha1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci U S A 105: 16242-16247. [Crossref]
- Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, et al. (2005) Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med 202: 885-891. [Crossref]
- Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, et al. (2005) Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A 102: 419-424. [Crossref]
- Jarnicki AG, Lysaght J, Todryk S, Mills KH (2006) Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol 177: 896-904. [Crossref]
- Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357: 539-545. [Crossref]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549-555. [Crossref]
- Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7: 211-217. [Crossref]
- Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A (2013) Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol 228: 1404-1412. [Crossref]
- Allavena P, Sica A, Solinas G, Porta C, Mantovani A (2008) The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol 66: 1-9. [Crossref]
- Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, et al. (2013) The role of IL-1β in the early tumor cell-induced angiogenic response. J Immunol 190: 3500-3509. [Crossref]
- Wu H, Xu JB, He YL, Peng JJ, Zhang XH, et al. (2012) Tumor-associated macrophages promote angiogenesis and lymphangiogenesis of gastric cancer. J Surg Oncol 106: 462-468. [Crossref]
- Chen P, Huang Y, Bong R, Ding Y, Song N, et al. (2011) Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin Cancer Res 17: 7230-7239. [Crossref]
- Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, et al. (2000) Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol 164: 762-767. [Crossref]
- Balkwill FR, Mantovani A (2012) Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol 22: 33-40. [Crossref]
- Mantovani A Biswas SK, Galdiero MR, Sica A, Locati M (2013) Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol 229: 176-185. [Crossref]
- Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122: 787-795. [Crossref]
- Perlino E, Cortese R, Ciliberto G (1987) The human alpha 1-antitrypsin gene is transcribed from two different promoters in macrophages and hepatocytes. EMBO J 6: 2767-2771. [Crossref]
- van 'tWout EF, van Schadewijk A, Savage ND, Stolk J, Hiemstra PS (2012) α1-antitrypsin production by proinflammatory and antiinflammatory macrophages and dendritic cells. Am J Respir Cell Mol Biol 46: 607-613. [Crossref]
- Serban KA, Petrusca DN, Lockett AD, Justice MJ, Petrache I (2013) Alpha 1 Antitrypsin Effect On Alveolar Macrophages Engulfment Of Apoptotic And Phagocytic Targets. C68. AUTOPHAGY AND APOPTOSIS: NEW FEATURES OF CELL FATE. American Thoracic Society International Conference Abstracts, (American Thoracic Society): A4766-A4766.
- Abecassis A, Schuster R, Shahaf G, Ozeri E, Green R,et al. (2014) alpha1-antitrypsin increases interleukin-1 receptor antagonist production during pancreatic islet graft transplantation. Cell Mol Immunol 11: 377-386. [Crossref]
- Schouppe E, De Baetselier P, Van Ginderachter JA, Sarukhan A (2012) Instruction of myeloid cells by the tumor microenvironment: Open questions on the dynamics and plasticity of different tumor-associated myeloid cell populations. Oncoimmunology 1: 1135-1145. [Crossref]
- Hurwitz AA, Watkins SK (2012) Immune suppression in the tumor microenvironment: a role for dendritic cell-mediated tolerization of T cells. Cancer Immunol Immunother 61: 289-293.[Crossref]
- Fridman WH, Galon J, Pagès F, Tartour E, Sautès-Fridman C, et al. (2011) Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res 71: 5601-5605. [Crossref]
- Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420: 860-867. [Crossref]
- Smith HA, Kang Y (2013) The metastasis-promoting roles of tumor-associated immune cells. J Mol Med (Berl) 91: 411-429. [Crossref]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, et al. (2004) NF-kappa B functions as a tumour promoter in inflammation-associated cancer. Nature 431: 461-466. [Crossref]
- Lockett AD, Kimani S, Ddungu G, Wrenger S, Tuder RM, et al. (2013) α₁-Antitrypsin modulates lung endothelial cell inflammatory responses to TNF-α. Am J Respir Cell Mol Biol 49: 143-150. [Crossref]
- Stetler-Stevenson WG, Aznavoorian S, Liotta LA (1993) Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol 9: 541-573. [Crossref]
- Zheng H, Takahashi H, Murai Y, Cui Z, Nomoto K, et al. (2006) Expressions of MMP-2, MMP-9 and VEGF are closely linked to growth, invasion, metastasis and angiogenesis of gastric carcinoma. Anticancer Res 26: 3579-3583. [Crossref]
- John A, Tuszynski G (2001) The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol Oncol Res 7: 14-23. [Crossref]
- Kleiner DE, Stetler-Stevenson WG (1999) Matrix metalloproteinases and metastasis. Cancer Chemother Pharmacol 43: S42-51. [Crossref]
- Joyce JA (2005) Therapeutic targeting of the tumor microenvironment. Cancer Cell 7: 513-520. [Crossref]
- Joyce JA, Pollard JW (2009) Microenvironmental regulation of metastasis. Nat Rev Cancer 9: 239-252. [Crossref]
- Sato N, Sutani A, Oya H, Yamaguchi T, Saito K, et al. (2007) Prognostic significance of neutrophil elastase inhibitor in patients with acute lung injury and interstitial pneumonia. Nihon Kokyuki Gakkaizasshi 45: 237-242. [Crossref]
- Inada M, Yamashita J, Nakano S, Ogawa M (1998) Complete inhibition of spontaneous pulmonary metastasis of human lung carcinoma cell line EBC-1 by a neutrophil elastase inhibitor (ONO-5046.Na). Anticancer Res 18: 885-890. [Crossref]
- Houghton AM (2013) Mechanistic links between COPD and lung cancer. Nat Rev Cancer 13: 233-245. [Crossref]
- DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, et al. (2009) CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16: 91-102. [Crossref]
- Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, et al. (2009) IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 206: 1457-1464. [Crossref]
- Mohamed MM, Cavallo-Medved D, Rudy D, Anbalagan A, Moin K, et al. (2010) Interleukin-6 increases expression and secretion of cathepsin B by breast tumor-associated monocytes. Cell Physiol Biochem 25: 315-324. [Crossref]
- Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124: 263-266. [Crossref]
- Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, et al. (2004) Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 104: 397-401. [Crossref]
- Jurasz P, Alonso-Escolano D, Radomski MW (2004) Platelet--cancer interactions: mechanisms and pharmacology of tumour cell-induced platelet aggregation. Br J Pharmacol 143: 819-826. [Crossref]
- Nash GF, Turner LF, Scully MF, Kakkar AK (2002) Platelets and cancer. Lancet Oncol 3: 425-430. [Crossref]
- Gans H, Tan BH (1967) Alpha-1-antitrypsin, an inhibitor for thrombin and plasmin. Clin Chim Acta 17: 111-117. [Crossref]
- Cho HJ, Jung JI, Lim do Y, Kwon GT, Her S,et al. (2012) Bone marrow-derived, alternatively activated macrophages enhance solid tumor growth and lung metastasis of mammary carcinoma cells in a Balb/C mouse orthotopic model. Breast Cancer Res 14: R81. [Crossref]
- Gil-BernabéAM, Ferjancic S, Tlalka M, Zhao L, Allen PD, et al. (2012) Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 119: 3164-3175. [Crossref]
- Yan HH, Pickup M, Pang Y, Gorska AE, Li Z, et al. (2010) Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res 70: 6139-6149.[Crossref]
- Lu X, Kang Y (2009) Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J Biol Chem 284: 29087-29096. [Crossref]
- Janciauskiene S, Larsson S, Larsson P, Virtala R, Jansson L,et al. (2004) Inhibition of lipopolysaccharide-mediated human monocyte activation, in vitro, by alpha1-antitrypsin. Biochem Biophys Res Commun 321: 592-600. [Crossref]
- Bellacen K, Kalay N, Ozeri E, Shahaf G, Lewis EC (2013) Revascularization of pancreatic islet allografts is enhanced by α-1-antitrypsin under anti-inflammatory conditions. Cell Transplant 22: 2119-2133. [Crossref]
- Stockley RA, Turner AM (2014) α-1-Antitrypsin deficiency: clinical variability, assessment, and treatment. Trends Mol Med 20: 105-115. [Crossref]