Multiple myeloma (MM) is consistently preceded by precursor states of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). These represent a continuum of progression of the tumor burden from the absence of symptoms or signs of end-organ damage towards full-blown symptomatic disease. MGUS, by definition presenting by monoclonal gammopathy without end organ damage, in fact might be associated with the numerous end organ lesions, first of all pathologic renal conditions. The term monoclonal gammopathy of renal significance (MGRS) was proposed by International Kidney and Monoclonal Gammopathy Research Group in order to discriminate the pathologic nature of these diseases from the truly benign MGUS. Spectrum of MGRS, caused by the deposition of monoclonal immunoglobulin’s or fragments thereof as organized and non-organized deposits, includes more commonly AL amyloidosis, monoclonal immunoglobulin deposition disease (MIDD) and light-chain proximal tubulopathy (LCPT). Same variants are seen in patients with MM and SMM. We present a rare case of combined immunoglobulin G kappa nephropathy: MIDD and proximal tubulopathy in a patient, manifested with acute kidney injury and diagnosed with SMM after 6 years of scrutinous repeated evaluation.
acute kidney injury, paraproteinemic nephropathy, kidney biopsy, monoclonal light and heavy chains
Multiple myeloma (MM) is a plasma-cell dyscrasia presenting with generalized neoplastic changes in bones, accompanied by impaired haematopoiesis and susceptibility to infections. The diagnosis is based on histologic, serologic, and radiographic features: bone marrow clonal plasma cells; monoclonal protein in the serum or urine; and end-organ damage, evidenced by renal impairment, hypercalcemia, anemia, or lytic bone lesions. Monoclonal gammopathy of undermined significance (MGUS) is a pre-malignant disorder, characterized by presence of monoclonal gammopathy without end organ damage. MGUS tend to progress over time to MM, lymphoproliferative disorders and AL amyloidosis with the rate about 1% per year. Asymptomatic or smoldering multiple myeloma (SMM) is a heterogeneous clinical entity where a subset of patients has an indolent course that mimics MGUS, whereas others have a more aggressive course that has been described as “early myeloma”. MM is consistently preceded by precursor states of MGUS and SMM, these represent a continuum of progression of the tumor burden from the absence of symptoms or signs of end-organ damage towards full-blown symptomatic disease [1-8].
Renal damage in MM is mainly caused by the deposition of monoclonal immunoglobulin’s (Ig) or fragments thereof as organized (casts, crystals, fibrils, microtubules) and non-organized deposits, involving all compartments of the renal parenchyma: glomeruli, tubules, interstitial space and vessels. Organized deposits induce cast-nephropathy, light-chain proximal tubulopathy (LCPT), AL amyloidosis, glomerulonephritis with organized microtubular monoclonal deposits (GOMMID), and cryoglobulinemic glomerulonephritis; while non-organized deposits lead to the monoclonal immunoglobulin deposition disease (MIDD) or proliferative glomerulonephritis with monoclonal deposits of IgG/IgA (PGNMID). It appears that amino acid sequence of the monoclonal light chains (LC) and other monoclonal proteins, defining inherent biochemical properties, is the primary determinant of the specific pattern of renal parenchymal deposition and clinical disease. Differential diagnostics demands pathology evaluation of kidney tissue, as the above mentioned lesions cannot be differentiated solely on the base of clinical presentation, which vary from acute kidney injury (AKI) and chronic kidney disease (CKD) to nephrotic syndrome (NS) and renal tubular disturbances [9-22].
MGUS, by definition presenting by monoclonal gammopathy without end organ damage, in fact might be associated with numerous end organ lesions, first of all pathologic renal conditions. To describe this paradox terms like “MIDD with MGUS” or “Glomerulonephritis with MGUS” have been used in the literature. It was shown that such conditions are associated with a high morbidity and mortality [23-33], and the term “monoclonal gammopathy of renal significance” (MGRS) was proposed by International Kidney and Monoclonal Gammopathy Research Group in order to discriminate the pathologic nature of these diseases from the truly benign MGUS [34].
Spectrum of MGRS includes more commonly AL amyloidosis, MIDD and LCPT, and rarely - PGNMID, GOMMID, cryoglobulinemic glomerulonephritis and AH amyloidosis. MGRS conditions are diagnosed by demonstration of monoclonal deposits in the kidney, therefore kidney tissue immunofluorescence/immunohistochemistry study and serum and urine monoclonal protein studies should be performed to match the monoclonal protein in circulation with the monoclonal deposits in the kidney. As MGRS is associated with high morbidity due to the severity of renal and sometimes systemic lesions induced by the monoclonal proteins, early recognition is crucial, as suppression of paraprotein secretion by chemotherapy often improves outcomes [35-39].
Here we present a case, manifested with AKI and diagnosed with SMM after 6 years of scrutinous repeated evaluation (summary of work-up and treatment is shown in the table 1).
Caucasian lady, 1945 year of birth. Primary admission to Nephrology, Botkin Memorial Hospital in May 2011.
Main complains
General weakness.
Previous medical history
Breast cancer with mastectomy, hysterectomy, ovariectomy and chemotherapy in 1999, oncologist follow-up for 10 years with no signs or symptoms of cancer recurrence and normal routine labs; mild arterial hypertension.
History of present illness
December 2010 she suddenly developed “dark” urine, within 3 days followed by anuria and was admitted to local hospital with BP 200/100 mm Hg and serum creatinine 800µmol/L. She was diagnosed with AKI and started on hemodialysis (HD), after 4 HD sessions her urine output restored, creatinine decreased to 200µmol/L and she was discharged.
Few days later she developed high grade fever with the second episode of anuria, and was admitted to local nephrology unit with creatinine 550µmol/L, Hb 10.0g/dL, proteinuria 5g/24 hours, microhematuria 10-20 RBC/hpf, and re-started on HD. Serum protein electrophoresis revealed M-band, serum and urine immunoelectrophoresis with Freelite assay found monoclonal serum IgG κ 8.4g/L and traces of the monoclonal IgG κ and LC κ in the urine.
She was suspected with multiple myeloma and referred to hematology. Her serum calcium was normal, skeletal X-ray did not reveal any destructive lesions, chest and abdomen CT was unremarkable. Bone marrow smear showed normal hematopoietic indices, plasma cells 1.5%, lymphocytes 6%.
Light microscopy: normal ratio of all three hematopoietic lineages with single plasma cells and small lymphoid cells. Congo red staining negative. Immunohistochemistry: the small proportion of dispersed lymphoid cells are CD79, CD20 and CD3 positive; single plasma cells, located interstitially and surrounding adipose droplets are CD38, CD138 positive. CD 56 positive cells not found. Pathologist’s conclusion: non-specific changes.
Before bone marrow biopsy reading became available patient was started on i.v. cyclophosphamide and oral prednisone. Within 3 weeks her urine output restored, creatinine decreased to 224 µmol/L, HD was discontinued. Her serum IgG κ decreased to 5.3 g/L, paraprotein in the urine was not found.
Only pathologist’s conclusion available: Diffuse immune-complex proliferative glomerulonephritis.
The diagnosis of MM was ruled out and chemotherapy (total dose of cyclophosphamide reached 4000 mg) was discontinued. She was diagnosed with “Chronic glomerulonephritis with MGUS” and referred to our clinic.
At admission
Conscious, alert, body temperature 36.7°C, RR 16 per minute, pulse regular 80 per minute, BP 160/90 mm Hg. Obese, skin normally coloured, scars after left mastectomy, hysterectomy. Mild pedal oedema. HEENT and neck otherwise normal. Peripheral lymph nodes not felt. Lungs: clear. Heart: regular rhythm, no murmur. Abdomen: soft, non-tender, bowel sounds normal. Liver, spleen and kidneys not felt. Urine normally coloured, urine output 1600 ml/24 hours.
Work-up
Proteinuria 0.2-0.03 g/l, Hb 11.4 g/dL, creatinine 143 µmol/L, uric acid 567 µmol/L, total calcium 2.34 mmol/L. IgG κ secretion 7.7 g/L. Bone marrow smear again sowed normal hematopoietic indices, plasma cells 1.5%, lymphocytes 7.5%.
Light microscopy: sections of formalin fixed paraffin-embedded tissue stained with H&E, Masson’s trichrome and periodic acid-Shiff. 12 glomeruli, 1 totally sclerotic, the rest are enlarged without any other changes. Interstitial fibrosis and tubular atrophy up to 20% of parenchyma. Few tubules contain РAS-positive casts without giant cell reaction; some of the casts are fragmented. Focal intensive monomorphic lymphoid interstitial infiltration. Arteries are not presented, arterioles without any changes. Immunofluorescence on unfixed frozen sections: negative for IgG, IgA, IgM, C3, Clq, κ and λ LC, and fibrinogen. Pathologist’s conclusion: Chronic interstitial nephritis. Re-evaluation of kidney biopsy in hematopathology unit: interstitial nephritis, no data for specific lymphoid infiltration.
Patient was diagnosed with “Chronic interstitial nephritis with MGUS”.
Further course
During next year she was treated for arterial hypertension and hyperuricemia in the outpatient setting. In May 2012 she developed anuria; at admission to our clinic serum creatinine was 621µmol/L, Hb 8.6g/dL, proteinuria 0.4g/24 hours, RBC 10-15 hpf. Further increase of IgG κ secretion to 11.0g/L was found (Table 1), repeated skeletal X-ray and chest CT were unremarkable. She was again started on HD, her urine output recovered, creatinine gradually decreased to 161µmol/L.
Table 1. Serum and urine immunoelectrophoresis, serum creatinine, diagnostic procedures and treatment data for 2010-2017
Date |
Serum IgG κ (g/L) |
Serum LC κ (mg/L) |
Urine IgG κ |
Urine LC κ |
Serum creatinine (µmol/L) |
Kidney biopsy |
Bone marrow biopsy |
HD |
Chemotherapy |
12.2010 |
8.4 |
- |
+ |
+ |
800-200 |
|
|
+ |
- |
01.2011 |
|
|
|
|
550-224 |
|
+ |
+ |
+ |
04.2011 |
5.3 |
- |
- |
- |
143 |
+ |
|
|
- |
07.2011 |
7.7 |
- |
- |
- |
188 |
+ |
|
|
- |
05.2012 |
11.0 |
- |
- |
- |
621-161 |
+ |
+ |
+ |
+ |
07.2012 |
9.9 |
- |
- |
- |
160 |
|
|
|
+ |
01.2013 |
12.5 |
- |
- |
- |
159 |
|
|
|
+ |
08.2013 |
13.3 |
181 |
- |
- |
163 |
|
+ |
|
+ |
10.2013 |
14.1 |
48 |
- |
- |
148 |
|
|
|
- |
08.2014 |
12.7 |
49 |
- |
- |
151 |
|
|
|
- |
01.2015 |
14.2 |
96 |
- |
- |
146 |
|
|
|
- |
07.2015 |
15.8 |
50 |
- |
- |
152 |
|
|
|
- |
09.2015 |
14.3 |
69 |
- |
- |
149 |
|
|
|
- |
09.2016 |
15.6 |
225 |
- |
+ |
178 |
|
+ |
|
- |
11.2016 |
|
|
|
|
163 |
|
|
|
+ |
12.2016 |
|
|
|
|
154 |
|
|
|
+ |
01.2017 |
15.0 |
114 |
- |
+ |
147 |
|
|
|
+ |
Light microscopy: sections of formalin fixed paraffin-embedded tissue stained with H&E, Masson’s trichrome, periodic acid-Shiff and Jones’ silver. Both cortex and medulla are presented, 25 glomeruli, including 4 (16%) completely sclerotic ones. The rest glomeruli are significantly enlarged, with mainly single-contoured walls; with diffuse and segmental mild mesangial widening due to PAS-positive extracellular matrix and mild mesangial hypercellularity; with focal and segmental minimal endocapillary hypercellularity, presented by few mononuclear cells in the capillary lumen. Masson’s trichrome stain shows diffuse irregular fuchsinophilic staining of mesangial matrix, Jones’ stain reveals focal and segmental double-contoured glomerular basement membrane (GBM). 2 glomeruli (8%) have nonspecific segmental sclerosis. There is no sign of crescentic lesion. (Figures 1-4). Tubular epithelial cytoplasm diffusely loaded with PAS-negative, Jones-negative (non-argyrophylic) protein droplets. Diffuse and focal acute tubular epithelial injury presented by loss of brush border and flattening of cell lining. Tubular lumens contain single small casts without specific tinctorial properties. Tubular basement membranes (TBM) of preserved non-atrophic tubules strongly PAS-positive, emphasized or focally moderately thickened, without multilayering or wrinkling. Masson’s trichrome stain shows multifocal fuchsinophilic segments in thickened tubular basement membranes. (Figures 5-10). Mild focal (20%) tubular atrophy with thickening, wrinkling and multilayering of TBM. Focal mild interstitial fibrosis (20%). Small size artery and arteriole walls severely thickened due to smooth muscle cell hypertrophy. Middle size artery walls are significantly thickened due to intimal fibrosis. Immunofluorescence on pronase-digested paraffin-embedded tissue fixed in formalin with FITC-conjugated anti IgA, IgG, IgM, C1q, C3, fibrinogen, κ and λ LC antibodies (using pronase antigen retrieval protocol): diffuse linear expression of IgG++/+++ and κ++/+++ along TBMs and GBMs. Fine granular confluent IgG++ mesangial expression. Diffuse expression of IgG++/+++ and κ+++ expression in the tubular epithelial cytoplasmic droplets. (Figures 11-16). Presented small cast sow equal expression of both κ+++ and λ+++. Electron microscopy: Single segments of subendothelial fine granular increase of GBM electron density and segmental subepithelial increase of TBM electron density (Figures 17,18). Pathologist’s conclusion: Combined monoclonal (IgG/κ) paraproteinemic nephropathy – MIDD with glomerulomegaly and mild mesangial hypercellulatity; and proximal paraproteinemic tubulopathy with acute tubular necrosis. Nonspecific secondary global (16%) and focal segmental (8%) glomerulosclerosis, mild interstitial fibrosis (20%); prominent arteriole-arteriosclerosis.
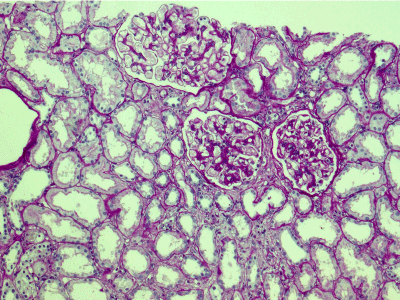
Figure 1. Enlarged glomeruli; mild mesangial widening and minimal endocapillary hypercellularity; acute tubular epithelial injury. PAS x 100.
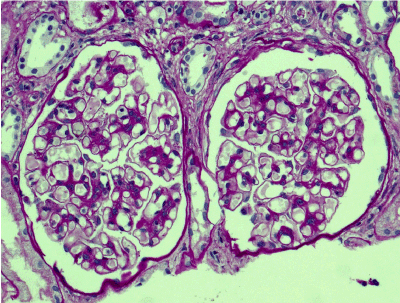
Figure 2. Enlarged glomeruli; mild mesangial widening and hypercellularity; segmental double-contoured capillary walls. PAS x 200.
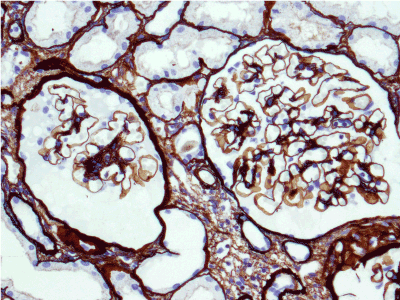
Figure 3. Enlarged glomeruli; mild segmental mesangial widening and hypercellularity; glomerular basement membrane single-contoured, regularly argyrophilic. Jones’ silver x 200.
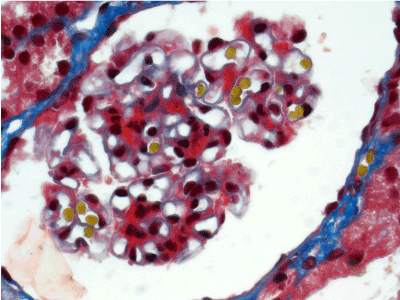
Figure 4. Fuchsinophilic staining of mesangial matrix. Masson’s trichrome х 400.
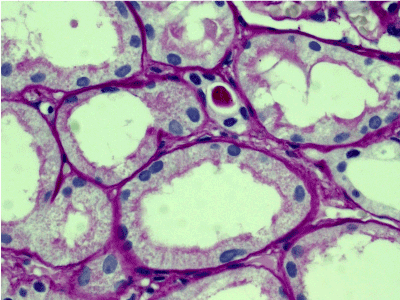
Figure 5. Convolute tubules epithelial cytoplasm with mainly preserved brush border, loaded with PAS-negative protein droplets. PAS x 400.
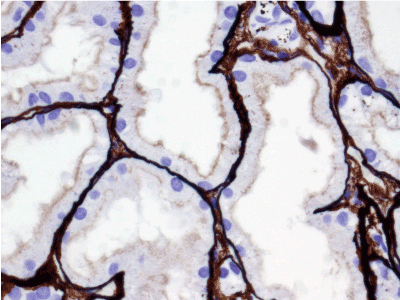
Figure 6. Convoluted tubules epithelial cytoplasm with mainly preserved brush border, filled with Jones-negative protein droplets. Jones’ silver x 400.
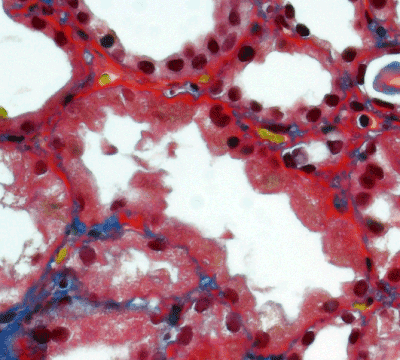
Figure 7. Thickened, irregular fuchsinopilic basement membranes of convoluted and straight tubules. Masson’s trichrome х 400.
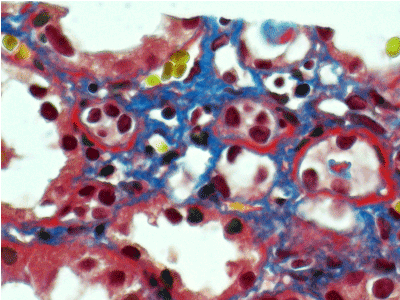
Figure 8. Thickened fuchsinopilic basement membranes of straight tubules. Masson’s trichrome х 400.
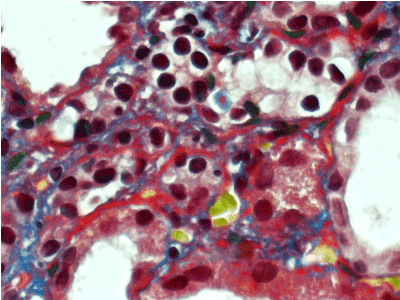
Figure 9. Thickened, irregular fuchsinopilic basement membranes of convoluted and straight tubules. Masson’s trichrome х 600.
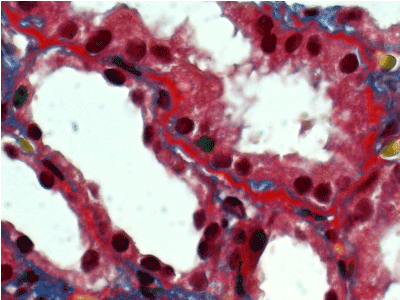
Figure 10. Thickened fuchsinopilic basement membranes of straight tubules. Masson’s trichrome х 600.
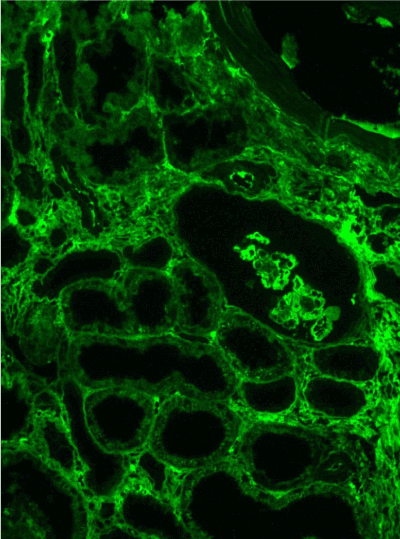
Figure 11. Diffuse linear expression of IgG along tubular and glomerular basement membranes. Immunofluorescence on formalin fixed paraffin-embedded sections (using pronase antigen retrieval protocol) х 100.
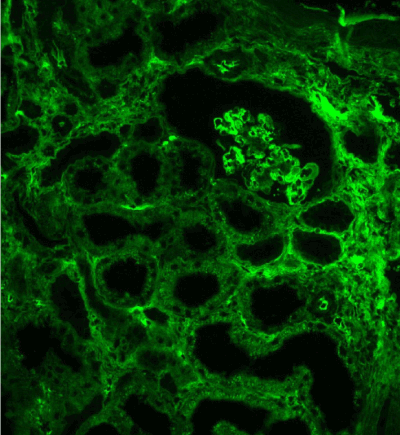
Figure 12. Diffuse linear expression of light chain κ along tubular and glomerular basement membranes. Immunofluorescence on formalin fixed paraffin-embedded sections (using pronase antigen retrieval protocol) х 100.
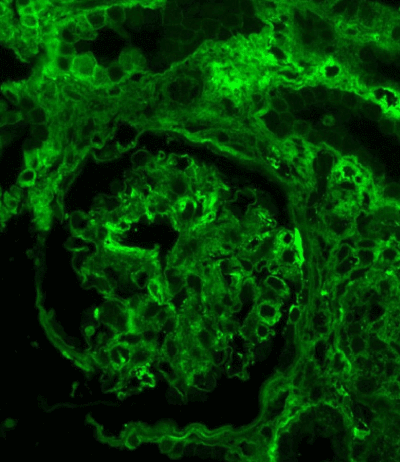
Figure 13. IgG expression in the glomerulus: linear along glomerular basement membrane and irregular granular-confluent in mesangium. Immunofluorescence on formalin fixed paraffin-embedded sections (using pronase antigen retrieval protocol) х 200.
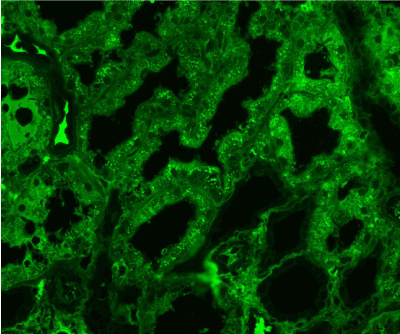
Figure 14. Diffuse IgG expression in the protein droplets in the tubular epithelial cytoplasm. Immunofluorescence on formalin fixed paraffin-embedded sections (using pronase antigen retrieval protocol) х 200.
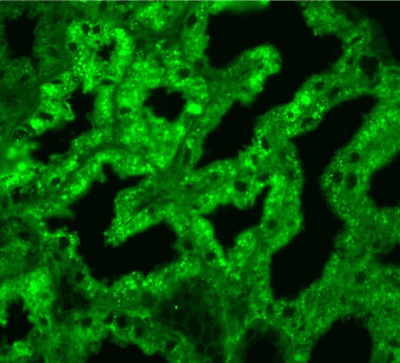
Figure 15. Diffuse light chain κ expression in the protein droplets in the tubular epithelial cytoplasm. Immunofluorescence on formalin fixed paraffin-embedded sections (using pronase antigen retrieval protocol) х 200.
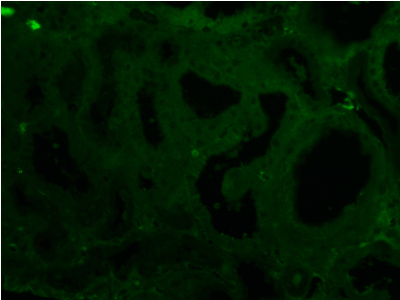
Figure 16. Negative light chain λ in any compartment of kidney tissue. Immunofluorescence on formalin fixed paraffin-embedded sections (using pronase antigen retrieval protocol) х 200.
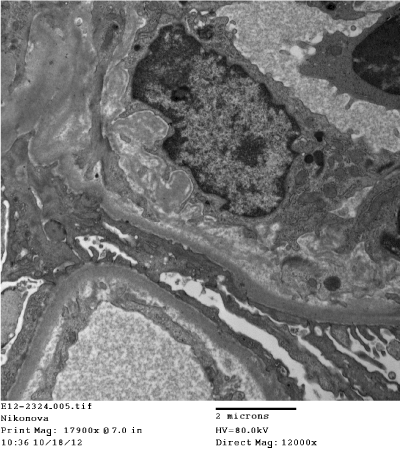
Figure 17. Segmental subendothelial fine granular increase of electron density of glomerular basement membrane. Electron microscopy x 12000
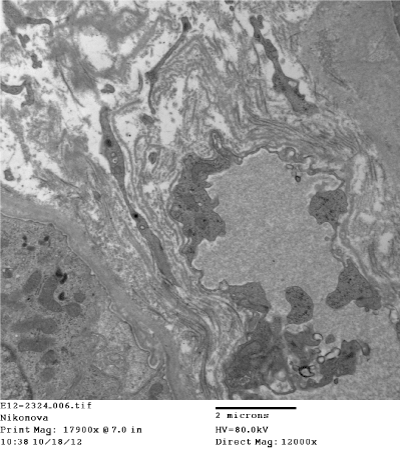
Figure 18. Segmental subepithelial increase of electron density of tubular basement membrane. Electron microscopy x 12000
Light microscopy: bone trabecules with focal resorption. Stromal repletion with focal hemorrhages. Bone marrow cavities are wide, bone marrow moderately cellular (with respect to normal for patient’s age). Megakaryocyte lineage is sufficient, megakaryocytes of relatively small size with hypolobular nuclei. Erythroid lineage is sufficient, erythrokariocytes of normoblastic type. Granulocytic lineage is sufficient, granulocytes of different degree of maturation, predominantly mature. Discrete small lymphoid cells are located interstitially; mature plasma cells are located perivascular. Pathologist’s conclusion: non-specific changes.
Patient was diagnosed with MGRS (MIDD IgG κ and proximal tubulopathy IgG κ), and July 2012 re-started on chemotherapy with i.v. cyclophosphamide plus dexamethasone - 5 day courses every 3 months (total dose of cyclophosphamide 8000 mg), under regular control of monoclonal secretion. 4-th course of chemotherapy was completed August 2013; serum creatinine was 148µmol/L, proteinuria 0.2g/24 hours. IgG κ secretion remained 14.1g/L; LC κ secretion 181mg/L, which appeared before the last chemotherapy course, decreased 48mg/L. Moderate decrease of polyclonal IgG was found.
Pathologist’s conclusion: No data in favor of lymphoproliferative disease.
She was doing well and followed-up in the outpatient setting for next 3 years. September 2016 her IgG κ and LC κ serum levels increased to 15.6g/L and 225mg/L respectively, and LC κ appeared in the urine. She was re-admitted to our clinic with Hb 12.3g/dL, creatinine 178µmol/L, uric acid 484µmol/L, total protein 8.6g/dL, total serum calcium 2.31mmol/L, proteinuria 0.2g/24 hours and urine RBC 8-10 hpf.
Light microscopy: Bone trabecules with focal resorption. Bone marrow cavities are wide, bone marrow is moderately cellular (with respect to normal for patient’s age). Granulocytic lineage is moderate, represented by cells of different maturation degree, predominantly mature. Erythroid lineage is moderate, represented by clusters of normoblastic erythrokariocytes, focally rejuvenated. Sufficient number of megakaryocytes is located discretely, represented by of relatively small size cells with hypersegmented normochromic nuclei. Discrete small lymphoid cells and mature plasma cells are located interstitially. Immunohistochemistry with anti CD3, CD19, CD20, CD56, CD138, IgA, IgG, light chains κ, light chains λ and CyclinD antibodies. Increased number of mature plasma cells (CD138+), located discreetly and in clusters perivascular, intra- and para-trabecular, and expressing CD56 (membrane reaction) and IgG (cytoplasmic reaction). They are predominantly κ-positive with only single of them λ-positive. Small B-cells are CD19 and CD20 positive, some plasma cells, located discreetly intra-trabecular are CD19 positive. Small B-cells (CD20+) and small T-cells (CD3+) are dispersed interstitially. Histiocytes are CyclinD positive. Pathologist’s conclusion: bone marrow pathology demonstrates moderate plasma cell infiltration with aberrant plasma cell immunophenotype IgG κ+, which can be characterized as minimal features of multiple myeloma.
Final diagnosis
Smoldering multiple myeloma with combined paraproteinemic kidney damage: MIDD IgG κ and proximal tubulopathy IgG κ, CKD stage 3b.
Current treatment and follow-up
Patient was referred to the hematology unit and November 2016 was started on bortezomib-cyclophosphamide-dexamethasone. At the latest follow-up visit January 25 2017, after 3 courses of chemotherapy she is doing well, her LC κ secretion decreased to 114mg/L, and serum creatinine - to 147µmol/L. Next course is scheduled on February 2017.
First clinical manifestation in our patient was anuric AKI of unexplained origin, partially resolved after few HD sessions. During the second episode of anuria, which developed shortly and demanded HD again, M-band was found by serum protein electrophoresis. Serum and urine immunoelectrophoresis revealed moderate monoclonal Ig κ secretion, and IgG κ and LC κ urinary excretion, therefore MM was suspected. Re-introduction of HD, followed by cyclophosphamide and prednisone treatment, lead to the restoration of kidney function, decrease of paraproteinemia and vanishing of paraproteinuria. However, her anemia was mild, serum calcium was normal with no evidence of lytic bone lesions, and bone marrow smear, as well as bone marrow biopsy did not confirm plasma cell or lymphocyte infiltration, and kidney biopsy did not show paraproteinemic kidney damage.
At that point MM was ruled out, and the diagnosis of “Chronic glomerulonephritis with MGUS” appeared on the basis of kidney biopsy conclusion. Of notice, 1-st biopsy was read by external pathologist and hardly can be interpreted. As this diagnosis was not compatible with two episodes of AKI, and monoclonal serum IgG κ increased again after chemotherapy discontinuation, we performed second kidney biopsy in search of LCPT, cast-nephropathy or specific lymphoid infiltration, missed by the first biopsy. Light microscopy showed interstitial nephritis with few РAS-positive casts without giant cell reaction, which could not prove cast-nephropathy, and just one area of intensive monomorphic lymphoid interstitial infiltration, which was not considered as specific. Immunofluorescence on unfixed frozen sections was totally negative; therefore, LCPT was also not confirmed. Hence, we did not find any glomerular lesions but very mild global glomerulosclerosis and moderate glomerulomegaly, and failed to prove any paraproteinemic kidney damage or lymphoid infiltration, we came to the formal diagnosis of “Interstitial nephritis with MGUS” and kept “watch and wait” strategy.
New episode of AKI happened after a year of follow-up, and further increase of monoclonal IgG κ secretion, forced us to perform the third kidney biopsy. Light microscopy showed glomerulomegaly, mild mesangial hypercellulatity and acute tubular necrosis. However, this time we used immunofluorescence on pronase-digested paraffin-embedded tissue, fixed in formalin [40], and found diffuse linear IgG++/+++ and κ++/+++ expression along TBM and GBM, fine granular confluent IgG++ mesangial expression, and diffuse IgG+++ and κ+++ expression in tubular epithelial cytoplasmic droplets. These findings confirmed paraproteinemic origin of renal damage and matched the monoclonal protein in circulation with the monoclonal deposits in the kidney. That led us to the diagnosis of combined paraproteinemic IgG κ nephropathy – MIDD and proximal paraproteinemic tubulopathy with acute tubular necrosis, the latter explained AKI episodes. The evidence for MM was still pending - repeated skeletal X-ray and bone marrow biopsy failed to confirm lytic bone changes and clonal plasma cells, therefore we arrived to the diagnosis of MGRS, the concept of which was just introduced [34].
Second block of chemotherapy resulted in the stabilization of kidney function – no new episodes of AKI occurred during next 3 years. However, IgG κ secretion was gradually increasing, and LC κ secretion appeared, but the third bone marrow biopsy again failed to give evidence for MM. Only substantial increase of LC κ secretion 3 years after the second chemotherapy guided us to perform forth bone marrow biopsy and order immunohistochemistry, which demonstrated moderate plasma cell infiltration with aberrant plasma cell immunophenotype IgG κ+ and allowed to end up with the diagnosis of SMM with combined paraproteinemic kidney damage. That was the sixth diagnosis during 6 years of follow-up and treatment (Table 2).
Table 2. List of diagnoses 2010-2016
Date |
Diagnosis |
12.2010 |
Acute kidney injury of unknown origin |
01.2011 |
Multiple myeloma |
04.2011 |
Glomerulonephritis with MGUS |
07.2011 |
Interstitial nephritis with MGUS |
05.2012 |
MGRS: monoclonal immunoglobulin deposition disease IgG κ and proximal tubulopathy IgG κ |
09.2016 |
Smoldering multiple myeloma with combined paraproteinemic kidney damage: monoclonal immunoglobulin deposition disease IgG κ and proximal tubulopathy IgG κ |
Analyzing the clinical course and pathology data we presume that if immunofluorescence on pronase-digested paraffin-embedded tissue had been available for the second kidney biopsy processing, and immunohistochemistry with the whole panel of antibodies had been done for the second or third bone marrow biopsy, the diagnosis of SMM with paraproteinemic kidney damage might have been already proven in 2011.
2021 Copyright OAT. All rights reserv
The coexistence of two and more types of paraproteinemic kidney damage, like amyloidosis and MIDD, cast-nephropathy and MIDD, cast-nephropathy, amyloidosis and MIDD, is described in the literature [41-43]; however, we did not find cases of combination of MIDD and LCPT.
On the other hand, paraproteinemic proximal tubular injury, including crystal-storing hystiocytosis and LCPT with or without Fanconi syndrome, and causative for acute tubular necrosis and AKI [13,18,22,37], is almost invariably associated with monoclonal LC κ, and only rarely with Ig heavy chains [44], like in our case. As no crystals were found by electron microscopy, and the patient did not demonstrate clinical features of Fanconi syndrome, we presume that she has non-crystalline form of proximal heavy and light chain tubulopathy without Fanconi syndrome, causing repeated episodes of AKI. Again, we couldn’t find such a pattern of injury in the available literature.
Of interest, at the time of the third biopsy, which was critical for the diagnosis, our patient had only monoclonal IgG κ secretion. LC κ were not detectable in the serum or urine either, which is compatible with the pathology findings, showing that the paraproteinemic kidney damage was IgG κ driven. We suppose that repeated kidney biopsy, if performed at the time when LC κ secretion appeared, might show also LC κ associated additional damage.
And finally, we believe that the chemotherapy with i.v. cyclophosphamide, which our patient received twice, even it was guided by incorrect diagnoses, was beneficial for our patient and delayed the progression both of blood disorder and CKD.
Unexplained kidney function disturbances in patients with monoclonal gammopathy demand usage of diagnostic algorithm, which include kidney biopsy with full-panel immunofluorescence, and bone marrow biopsy with full-panel imunohistochemistry. We consider very useful to apply immunofluorescent duplication on pronase-digested paraffin-embedded tissue especially in cases of discrepancy between clinical evidence of monoclonal secretion and confusing pathology findings. Patients with MGRS or SMM, diagnosed on the basis of above-mentioned work-up findings benefit from MM-protocols chemotherapy.
We have to thank doctors Olga Vinogradova, Vladimir Lunin, Sergey Semochkin, Alla Kovrigina, Salya Maryina and Arthur Cohen for their help in the diagnostics and treatment of our patient.
- International Myeloma Working Group (2003) Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: A report of the International Myeloma Working Group. Br J Haematol 121: 749-757. [Crossref]
- Rajkumar SV, Kyle RA (2005) Multiple myeloma: diagnosis and treatment. Mayo Clin Proc 80: 1371-1382. [Crossref]
- Korbet SM, Schwartz MM (2006) Multiple myeloma. J Am Soc Nephrol 17: 2533-2545. [Crossref]
- Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, et al. (2006) Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 354: 1362-1369. [Crossref]
- Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, et al (2009) Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood 113: 5412-5417. [Crossref]
- Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, et al (2010) Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 24: 1121-1127. [Crossref]
- Landgren O (2013) Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma: biological insights and early treatment strategies. Hematology Am Soc Hematol Educ Program 2013: 478-487. [Crossref]
- Ghobrial IM, Landgren O (2014) How I treat smoldering multiple myeloma. Blood 124: 3380-3388. [Crossref]
- Haas M, Spargo BH, Wit EJ, Meehan SM (2000) Etiologies and outcome of acute renal insufficiency in older adults: a renal biopsy study of 259 cases. Am J Kidney Dis 35: 433-447. [Crossref]
- Pozzi C, D’Amico M, Fogazzi GB, Curioni S, Ferrario F, et al (2003) Light chain deposition disease with renal involvement: clinical characteristics and prognostic factors. Am J Kidney Dis 42: 1154-1163. [Crossref]
- Markowitz GS (2004) Dysproteinemia and the kidney. Adv Anat Pathol 11: 49-63. [Crossref]
- Herrera GA, Joseph L, Gu X, Hough A, Barlogie B (2004) Renal pathologic spectrum in an autopsy series of patients with plasma cell dyscrasia. Arch Pathol Lab Med 128: 875-879. [Crossref]
- Herrera GA, Sanders PW (2007) Paraproteinemic renal diseases that involve the tubule-interstitium. In: Herrera GA (ed): The Kidney in Plasma Cell Dyscraisas. Contrib Nephrol. Basel, Karger 153: 105-115.
- Merlini G, Pozzi C (2007) Mechanisms of renal damage in plasma cell dyscrasias: an overview. Contrib Nephrol 153: 66-86. [Crossref]
- Batuman V (2007) Proximal tubular injury in myeloma. Contrib Nephrol 153: 87-104. [Crossref]
- Ronco P, Plaisier E, Aucouturier P (2010) Ig-related renal disease in lymphoplasmacytic disorders: an update. Semin Nephrol 30: 557-569. [Crossref]
- Heher EC1, Goes NB, Spitzer TR, Raje NS, Humphreys BD, et al. (2010) Kidney disease associated with plasma cell dyscrasias. Blood 116: 1397-1404. [Crossref]
- El Hanel C, Thierry A, Trouillas P, Bridoux F, Carrion C, et al. (2010) Crystal-storing histiocytosis with renal Fanconi syndrome: pathological and molecular characteristics compared with classical myeloma-associated Fanconi syndrome. Nephrol Dial Transplant 25: 2982-2990. [Crossref]
- Tsakiris DJ, Stel VS, Finne P, Fraser E, Heaf J, et al (2010). Incidence and outcome of patients starting renal replacement therapy for end stage of renal disease due to multiple myeloma or light chain depositon disease: an ERA-EDTA Registry study. Nephrol Dial Transplant 25: 1200-1206. [Crossref]
- Ronco P (2010) Disease classification: a pitfall of the ERA/EDTA registry? Nephrol Dial Transplant 25: 1022-1024. [Crossref]
- Davenport A, Merlini G (2012) Myeloma kidney: advances in molecular mechanisms of acute kidney injury open novel therapeutic opportunities. Nephrol Dial Transplant 27: 3713-3187. [Crossref]
- Cambier JF, Ronco P (2012) Onco-nephrology: glomerular diseases with cancer. Clin J Am Soc Nephrol 7: 1701-1712. [Crossref]
- Lin J, Markowitz GS, Valeri AM, Kambham N, Sherman WH, et al. (2001). Renal monoclonal immunoglobulin deposition disease: the disease spectrum. J Am Soc Nephrol 12: 1482-1492. [Crossref]
- Paueksakon P, Revelo MP, Horn RG, Shappell S, Fogo AB (2003) Monoclonal gammopathy: significance and possible causality in renal disease. Am J Kidney Dis 42: 87-95. [Crossref]
- Merlini G, Stone MJ (2006) Dangerous small B-cell clones. Blood 108: 2520-2530. [Crossref]
- Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E, et al. (2004). Proliferative glomerulonephritis with monoclonal IgG deposits; a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int 65: 85-96. [Crossref]
- Soares SM, Lager DJ, Leung N, Haugen EN, Fervenza FC (2006). A proliferative glomerulonephritis secondary to a monoclonal IgA. Am J Kidney Dis 47: 342-349. [Crossref]
- Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, et al. (2009) Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol 20: 2055-2064. [Crossref]
- Bridoux F, Hugue V, Coldefy O, Goujon JM, Bauwens M, et al (2002) Fibrillary glomerulonephritis and immunotactoid (microtubular) glomerulopathy are associated with distinct immunologic features. Kidney Int 62: 1764-1775. [Crossref]
- Nagao T, Okura T, Miyoshi K, Watanabe S, Manabe S, et al. (2005) Fibrillary glomerulonephritis associated with monoclonal gammopathy of undetermined significance showing lambda-type Bence Jones protein. Clin Exp Nephrol 9: 247-251. [Crossref]
- Sethi S, Zand L, Leung N, Smith RJ, Jevremonic D, et al. (2010) Membranoproliferative glomerulonephritis secondary to monoclonal gammopathy. Clin J Am Soc Nephrol 5: 770-782. [Crossref]
- Gertz MA, Leung N, Lacy MQ, Dispenzieri A, Zeldenrust SR, et al. (2009) Clinical outcome of immunoglobulin light chain amyloidosis affecting the kidney. Nephrol Dial Transplant 24: 3132-3137. [Crossref]
- Leung N, Buadi F, Song KW, Magil AB, Cornell LD, et al. (2010) A case of bilateral renal arterial thrombosis associated with cryocrystalglobulinaemia. Nephrol Dial Transplant Plus 3: 74-77. [Crossref]
- Leung N, Bridoux F, Hutchison CA, Nasr SH, Cockwell P, et al. (2012) Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood 120: 4292-4295. [Crossref]
- Fermand JP, Bridoux F, Kyle RA, Kastritis E, Weiss BM, et al. (2013) How I treat monoclonal gammopathy of renal significance (MGRS). Blood 122: 3583-3590. [Crossref]
- Cabrita A (2014) Monoclonal gammopathy of renal significance: why is it significant. J Blood Disord 1: 2-3.
- Paueksakon P, Fogo AB (2014) More light shed on light chains. Nephrol Dial Transplant 29: 1799-1801. [Crossref]
- Bridoux F, Leung N, Hutchison CA, Touchard G, Sethi S, et al (2015). International Kidney and Monoclonal Gammopathy Research Group. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int 87: 698-711. [Crossref]
- Leung N, Nasr SH (2016) A Patient with Abnormal Kidney Function and a Monoclonal Light Chain in the Urine. Clin J Am Soc Nephrol 11: 1073-1082. [Crossref]
- Nasr SH, Galgano SJ, Markowitz GS, Stokes MB, D'Agati VD (2006) Immunofluorescence on pronase-digested paraffin sections: a valuable salvage technique for renal biopsies. Kidney Int 70: 2148-2151. [Crossref]
- Gokden N, Cetin N, Colakoglu N, Kumar J, Abul-Ezz S, et al. (2007) Morphologic manifestations of combined light-chain deposition disease and light-chain cast nephropathy. Ultrastruct Pathol 31: 141-149. [Crossref]
- Qian Q, Leung N, Theis JD, Dogan A, Sethi S (2010) Coexistence of myeloma cast nephropathy, light chain deposition disease, and nonamyloid fibrils in a patient with multiple myeloma. Am J Kidney Dis 56: 971-976. [Crossref]
- Lorenz EC, Sethi S, Poshusta TL, Ramirez-Alvarado M, Kumar S, et al. (2010) Renal failure due to combined cast nephropathy, amyloidosis and light-chain deposition disease. Nephrol Dial Transplant 25: 1340-1343. [Crossref]
- Lebeau A, Zeindl-Eberhart E, Muller EC, Muller-Hocker J, Jundblut PR, et al. (2002) Generalized crystal-storing hystiocytosis associated with monoclonal gammopathy: molecular analysis of a disorder with rapid clinical course and review of literature. Blood 100: 1817-1827. [Crossref]