Despite advances in the treatment of acute myeloid leukemia (AML), patients still show high relapse and resistance against conventional chemotherapy. This resistance is related to a small clone referred to as Leukemia Stem Cells (LSCs). New targeted strategies are directed against the LSCs’ extrinsic regulators including their microenvironment such as a CXCR4 antagonist that is used to interfere with LSCs’ homing. Targeting LSCs’ surface molecules such as CD33 for selective elimination of LSCs has variable degrees of success that may require further assessments. Trials with CARs cells were effective in eradication of acute lymphoblastic leukemia, and they may have an effective role also in AML. Other strategies are directed against the intrinsic regulators such as self-renewal mechanisms and epigenetic reprogramming of LSCs. This review highlights targeting of the extrinsic regulators of the LSCs and identifies biological differences between them and normal hematopoietic stem cells.
acute myeloid leukemia, chimeric antigen receptors, hematopoietic stem cells, leukemic stem cells, extracellular targeting of acute myeloid leukemia
Acute myeloid leukemia (AML) is a hematological disorder characterized by a malignant clone thought to be derived from a small number of cells known as leukemic stem cells (LSCs). LSCs have a great ability for limitless self-renewal and also generation of leukemic progenitors that actively divide and produce a large number of blasts [1].
Most AML patients treated with conventional chemotherapies can achieve remission, but about 60% of remission patients still relapse. AML relapse may originate from LSCs (the chemo-resistant clone), and therefore targeting LSCs should be a main aim in the search for a cure for human AML. LSCs and normal hematopoietic stem cells (HSCs) share different properties and surface markers, so it is necessary to identify a signaling pathway that is specific to the AML LSC stage to eliminate them without damaging normal HSCs [2]. For targeting LSCs, it is important to overcome the properties that make LSCs resistant to therapy such as quiescence in the bone marrow niche and ability for limitless self-renewal [3].
Different trials have been performed to isolate LSCs, and it was reported that LSCs were present within the CD34+/CD38- clone; this clone is known to establish human AML in a xenograft model. In contrast, it has been observed that CD34- clones from the majority of nucleophosmin-1 (NPM-1) mutated AML are able to initiate human AML in a xenograft model. Thus, the isolation of LSCs on the basis of their phenotypic expression alone might not be enough [4].
In this review we present the newer therapeutic approaches that target the extrinsic LSC regulators, including manipulation of LSCs’ microenvironment. Cellular therapy depending on genetic engineering of T cells with chimeric antigen receptors (CARs) showed good results in pediatric acute lymphoblastic leukemia (ALL) and may have a role in the management of AML [5]. Other trials that utilized surface markers achieved different levels of elimination of AML [6].
LSCs are able to divide to progeny clonogenic blast cells, leading to the concept that AML is arranged in a hierarchy, with the LSCs present at the apex and the more “differentiated” blasts representing the main tumor bulk [7]. Some researchers argue that normal HSCs are the “cells of origin” for LSCs as a result of the similarity between normal and leukemic stem cells [8].
In general, LSCs widely differ in their self-renewal potential (long-term and short-term LSCs), demonstrating that the LSC still retains some aspects of normal HSC properties. This information supports the concept that HSCs are the cellular targets for transformations, not progenitors or the blast population. LSCs and HSCs share several properties that render them as stem cells, especially limitless self-renewal activities. With the discovery of the role of Bmi-1 as a key regulator for self-renewal mechanisms in LSCs, the stem cell model of leukemogenesis was given a strong boost [7].
Other researchers had argued that non-stem cells could be converted into LSCs if the correct self-renewal machinery were activated in these cells. Using retrovirus vectors with fusion oncogene MLL-AF9 in murine models provided evidence that the cell of origin of LSC may be a progenitor, related to the more phenotypically mature cells in comparison to the primitive HSC [9]. With regard to retroviral transformation in a murine model, LSC also did not express the stem cell antigen Sca1 and had an immune phenotype more related to normal granulocyte-macrophage progenitors (GMP) [7].
So from those two models, the transforming events (i.e., oncogenic mutations) may occur in either HSC (long-term HSC) or in progenitors (such as GMP) [8]. In relation to the long-term HSC, mutations may give rise to a pre-leukemic condition associated with expansion of HSCs and increase the genetic instability. Second hit (genetic event) will be needed to develop the full leukemic phenotype (Figure 1) [10]. However, mutations that occur within the progenitors may confer the property of limitless self-renewal to those cells (e.g., expression of the MLL-AF9 fusion oncogene), giving rise to the full leukemic phenotype (Figure 1) [8].
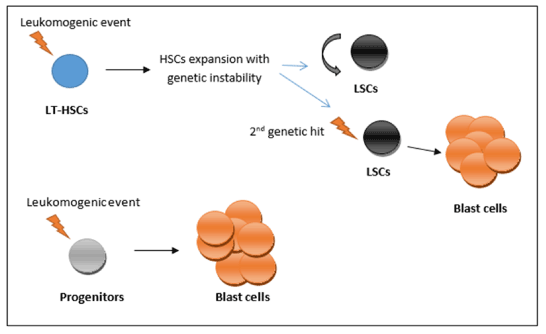
Figure 1. Leukemia stem cells (LSC) hierarchy
The transforming events (i.e., oncogenic mutations) may occur in either long-term hematopoietic stem cells (LT-HSC) or in progenitors (such as granulocyte-macrophage progenitors). In relation to the LT-HSC, mutations may give rise to a preleukemic condition associated with expansion of HSCs and increase the genetic instability. Second hit (genetic event) will be needed to develop the full leukemic phenotype. On the other hand, mutations that occur within the progenitors may confer the property of limitless self-renewal to those cells (e.g., expression of the MLL-AF9 fusion oncogene), giving rise to the full leukemic phenotype.
Bone marrow microenvironment is important for HSCs and LSCs. Two anatomical HSC niches that may have overlapping roles can be defined: the endosteal niche [11] and the perivascular niche [12]. Osteoblast, osteoclasts, and osteopontin are essential for regulation of HSC count and engraftment within the niche [13]. Osteoblast in endosteal niche provides essential support to HSCs because the manipulation of osteoblasts could increase HSC number [14]. Other observations support that abnormal osteoblast has no effect on HSCs. For example, the increase of the number of osteoblasts under treatment with strontium was not associated with effects on HSC numbers [15]. In contrast, the perivascular niche depends on cell/cell interactions. The interactions between chemokine receptor 4 (CXCR4) on HSCs’ surface and chemokine ligand 12 (CXCL12, also known as stromal-derived factor 1 alpha, SDF1-α) that expressed on mesenchymal stromal cells, and also the interactions between integrins (such as VLA-4) on HSCs and VCAM-1 on stromal cells [16] appear to be essential for HSCs’ homing [17].
The bone marrow microenvironment appears to be responsible for protecting LSCs by keeping them at quiescent state since many chemotherapeutic drugs target actively cycling cells [18]. LSCs close to the endosteal cells appear more quiescent and more resistant to the conventional chemotherapy drug cytarabine [18]. Manipulation of this microenvironment may be effective in the elimination of LSCs [19].
Targeting LSCs is the new hope to eradicate AML without harm to normal HSCs. It is important to overcome the properties that make LSCs resistant to therapy such as their ability for limitless self-renewal and quiescence in bone marrow niche [20].
Disrupting LSCs’ microenvironment
LSCs are able to receive and send several signals within bone marrow niche that play an important role in maintaining their quiescence. Several factors and signals such as Wnt/β-catenin and CXCL12/SDF1-α are involved in the relation between bone marrow microenvironment and LSCs. Also, the bone marrow niche is composed of both well vascularized zones and hypoxic zones that may affect LSCs’ activity [21].
LSCs have demonstrated over-expression of CXCR4, which is related to a poor prognosis. Inhibition of the CXCR4–SDF-1 interaction was noticed to decrease the engraftment of the Nalm-6 (ALL cell line), after the transplantation in mouse bone marrow [22].
AMD3465 is a CXCR4 antagonist that is used to interfere with CXCR4/SDF1-α and demonstrates a good effect in addition to conventional chemotherapy [6]. AMD3465 inhibits stromal cell-induced pro-survival signals in AML cells. Also, it was shown that AMD3465 increases the sensitivity of Flt-3-mutated AML cells to sorafenib (Flt-3 inhibitor). Both mobilization of AML cells into the peripheral circulation and the increased sensitivity to sorafenib decreased the AML burden in mice following sorafenib treatment [23]. AMD3100, another CXCR4 antagonist, was shown to increase the mobilization of murine acute promyelocytic leukemia cells. The combination of AMD3100 with chemotherapy diminished AML tumor burden and increased mice survival compared with chemotherapy alone [24].
Interaction between integrin VLA-4 on the LSCs and fibronectin on the stromal cells is associated with activation of the PI3K pathway (pro-survival) in AML cells. The effect of interfering with the interaction between VLA-4 and fibronectin and its role in sensitizing AML cells to drugs (e.g. cytosine arabinoside) has been demonstrated by administration of VLA-4 neutralizing antibodies that eliminate the relative resistance of VLA-4 (+) AML cells to drug-induced apoptosis [25]. The effector of the previous interactions, the Rac family (especially Rac1 and Rac2), also may be a target to inhibit LSCs’ homing in bone marrow niche. The Rac1 inhibitor NSC23766 has been found to succeed in delaying leukemia development in a mouse model [26].
Leukemic cells have been shown to express vascular endothelial growth factor (VEGF) and its receptor VEGFR-2. Recent studies illustrated the autocrine and paracrine functions of VEGF that mediate leukemic cell proliferation and the role of anti-VEGFR-2 in inhibition of leukemic propagation in a NOD/SCID xenograft model. Thus, VEGFR-2 signal targeting may be a promising strategy against AML in the near future [24].
Inhibition of LSC homing by blocking the interleukin-3 (IL-3) receptor has been tested. Ex vivo culture of LSCs with anti-IL-3 α chain neutralizing antibody, 7G3, significantly reduced the engraftment of transplanted AML cells in NOD/SCID mice, resulting in prolonged survival. However, transplantation of cord blood hematopoietic cells after co-culture with 7G3 did not affect the engraftment in comparison with AML-treated cells, which related to the significantly lower expression of IL-3 on CD34+CD38-HSCs compared with LSCs [27]. CD44, a hyaluronic acid receptor, is also one of the important hematopoietic stem and progenitor cell homing receptors. Treatment with an anti-CD44 antibody resulted in LSC mobilization in both a mouse model of chronic myelogenous leukemia and a xenograft model of human AML [28].
Another strategy depends on interfering with the ability of LSCs to thrive within the bone marrow microenvironment. Hypoxia inducible factor-1 alpha (HIF-1α) is a transcription factor that is induced in hypoxic conditions such as those typically found in the bone marrow microenvironment [6]. A HIF-1α inhibitor, echinomycin, was shown to eliminate leukemia stem cells in a xenograft transplant model without marked damage to the normal HSCs. Other HIF-1α inhibitors such as EZN-2968 and PX-478 are currently in clinical trials [29]. Other researchers have studied forced cell cycle induction by interferon-alfa (IFN-α) that will decrease LSCs’ resistance to treatment with chemotherapeutic agents such as 5-fluorouracil by overcoming their quiescence state [30].
Raaijmakers et al. demonstrated that deletion of certain bone marrow mesenchymal cells’ genes such as the Schwachmann–Bodian–Diamond syndrome gene led to development of myelodysplasia and AML in mice. These results strongly suggest that the development of certain hematologic malignancies cells may be derived by the transformation of bone marrow niche cells [31].
Targets expressed on the surface of AML LSCs (Monoclonal antibody)
CD33
Many trials have been designed to target CD33. The Gemtuzumab Ozogamicin (GO, Mylotarg®) trial tested a humanized anti-CD33 monoclonal antibody (mAb) conjugated with calicheamicin (DNA-damaging toxin) [32]. However, recently this drug was withdrawn because of its toxic effects on normal cells. The presence of CD33- LSCs and CD33+ normal HSCs has been reported; this observation lowers the therapeutic role of the anti-CD33 antibodies [33]. The remission rate can be increased with a combination of GO and valproic acid (epigenetic modifying drug) thereby facilitating the intercalation of calicheamicin with DNA, followed by induction of synergistic apoptosis (Table 1) [21].
CD123
Several trials to target CD123 have had successful results related to the mAb anti-CD123 (7G3) (Table 1) [27]. Use of fusion protein 26292(Fv)-PE38-KDEL, consisting of a CD123-specific single chain fragment of the variable regions (scFv) antibody that is fused to a pseudomonas exotoxin A fragment (PE38), had a potent apoptotic effect on different CD123+ leukemia cell lines [34]. Other 7G3-derived recombinant mAbs, CSL360 and CSL362, had a more potent effect [5]. The recombinant immunotoxin DT388IL3, consisting of a truncated diphtheria toxin fused to human IL-3 ligand, was studied but the clinical results were not promising [35]. The expression of CD123 on both normal HSCs and regenerating bone marrow was reported, so that the combination with other target markers may be safer [33].
CD44
CD44 plays an important role in engraftment of LSCs and HSCs [36]. Because the expression of CD44 is high in LSCs, targeting of CD44 (using the H90 mAb) might impair the LSCs’ engraftment in the bone marrow (Table 1) [28]. Jin et al. showed that treatment with H90 significantly increased survival of NOD/SCID mice transplanted with AML LSCs (CD34+CD38-) and reduced the LSC burden in the bone marrow of those mice. In contrast, the leukemic cells obtained from primary mice (H90 treated mice) failed to engraft into the secondary recipient mice [28]. Zhou and Chng explained the efficacy of the anti-CD44 mAb to eliminate LSC by three mechanisms: induction of leukemic cell differentiation, inhibition of cell proliferation, and disruption of LSCs’ homing [37].
CD47
CD47 is a transmembrane protein that interacts with signal regulatory protein (SIRPα) on phagocytes [38]. CD47 presents also in high expression in LSCs and has an important role in protection of LSCs from phagocytosis (Table 1). The interaction between CD47 of LSCs with SIRPα on phagocytes is a defense strategy (“Don’t eat me strategy”), enabling LSCs to escape phagocytosis, so targeting of CD47 has strongly enabled phagocytosis of LSCs [39]. Anti-CD47 mAbs have been used in xenograft mice models to block the CD47/SIRPα interaction, resulting in induction of phagocytosis of leukemic cells and eradication of LSCs [37]. The use of soluble SIRPα-Fc fusion proteins is another promising strategy, which leads to initiation of macrophages-mediated phagocytosis, resulting in a strong anti-leukemic effect and eradication of LSCs [40].
C-type lectin-like molecule-1
C-type lectin-like molecule-1 (CLL-1) has been found to be expressed in more than 87% of AML patients, while it is weakly expressed by normal hematopoietic cells [38]. For AML, both blast cells and LSCs express CLL-1 (86.5% vs. 54.5, respectively) (Table 1) [41]. However, the anti–CLL-1 antibody didn’t show the expected anti-leukemic effect, since anti-CLL-1 mAbs have been tested in vitro against the CLL-1+HL60 cells and had no effect on cell proliferation. Combination of a toxic moiety to the anti–CLL-1 antibody might be a promising targeting strategy to eradicate LSCs [42].
T cell immunoglobulin mucin-3
T cell immunoglobulin mucin-3 (TIM-3) is one of the most recent markers that has been found to be highly expressed in LSCs with no expression in normal HSCs (Table 1) [43]. Kikushige et al. demonstrated that TIM-3+AML population engrafted and initiated human AML in NRG mice, while TIM-3-AML population did not, suggesting that LSCs mostly present within the TIM-3+ population [44].
Anti-TIM-3 antibodies (ATIK2a) tested in mice models succeeded in blocking the human AML engraftment and development of leukemia in NOD/SCID mice, without any effect on normal HSC engraftment. Furthermore, ATIK2a treatment reduced or eliminated the re-engraftment of primary recipients’ bone marrow cells into secondary recipients [44].
FMS-related tyrosine kinase 3
FMS-related tyrosine kinase 3 (FLT-3) is one of the most frequent mutations in AML [45]. A neutralizing mAb specific for FLT3, named IMC-EB10, is effective in inhibiting growth and expansion of leukemic cells [46]. FLT-3 inhibitors such as lestaurtinib have been used in clinical trials involving patients with refractory AML, showing a great ability to induce rapid clearance of blasts from peripheral and bone marrow without associated normal cell toxicity [47]. Combined therapy of FLT-3 inhibitors with conventional chemotherapy was associated with about 92% complete remission [48].
Cellular therapy (Genetic engineering of T cells with chimeric antigen receptors)
Genetic engineering of T cells with chimeric antigen receptors (CARs) represents a revolution in targeted cellular therapies. The extracellular antigen-binding domain of CARs consists of the variable regions of the light and heavy chains of immunoglobulins, and the intracellular domain usually consists of the CD3 zeta chain of the T cell receptor complex (T cell activating domain). This unique combination increases efficacy of the engineered T cells against malignant cells [5]. For AML, a few CARs have been developed against different target molecules such as CARs specific for CD44v6 and CD123 [5].
Cytokine-induced killer cells, a natural killer like cells population, emerged as a novel therapy against chemo-resistant AML through the intrinsic non-specific antitumor activity of these cells [49]. Combination therapy of cytokine-induced killer cells and CARs showed a potent killing activity against leukemic cells, with a promising strategy for safe and effective eradication of LSCs [5].
There are many publications demonstrating various clinical trials of CAR cells in hematologic malignancies including AML by targeting Lewis-Y antigen (tumor-associated carbohydrate antigen on AML cells). One of those trials included 4 AML patients and resulted in improvement for 2 patients (1 transient reduction in blasts, 1 transient cytogenetic remission) [50].
For decades, researchers have been seeking to identify LSCs, the leukemic resistance clone that is responsible for initiation and recurrence of AML. Recent studies have shown that a high level of LSCs in patients is associated with poor prognosis such that effective eradication of these cells is needed in order to cure AML.
Different strategies are established based on understanding LSCs’ behaviors. Some researchers study and target the LSCs’ microenvironment, others study survival, growth, and apoptotic pathways, and other groups are interested in isolation of LSCs through different cell surface antigens such as CD33, CD123, CLL-1, CD44, CD47, and TIM-3 that are preferentially expressed on LSCs as compared to HSCs. But still the variability in expression of the target molecules and also the differences in response from patient to patient represent a major challenge for scientists, and further studies for the detection of the ideal weapon against this resistance clone are needed.
As we discussed in this review, until now there have been many trials with different levels of success because of the difference in expression of each target from one patient to another. Therefore, development of personalized therapies may be a promising way to design an ideal strategy for each patient.
References
- Hope KJ, Jin L, Dick JE (2004) Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol 5: 738-743. [Crossref]
- Anderson AC (2012) Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol 24: 213-216. [Crossref]
- Sands WA, Copland M, Wheadon H (2013) Targeting self-renewal pathways in myeloid malignancies. Cell Commun Signal 11: 33. [Crossref]
- Sarry JE, Murphy K, Perry R, Sanchez PV, Secreto A, et al. (2011) Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rgammac-deficient mice. J Clin Invest 121: 384-395. [Crossref]
- Tettamanti S, Magnani CF, Biondi A, Biagi E. (2013) Acute myeloid leukemia and novel biological treatments: monoclonal antibodies and cell-based gene-modified immune effectors. Immunol Lett 155: 43-46. [Crossref]
- Felipe Rico J, Hassane DC, Guzman ML (2013) Acute myelogenous leukemia stem cells: from Bench to Bedside. Cancer Lett 338: 4-9. [Crossref]
- Scatena R, Mordente A, Giardina B (2012) Advances in Cancer Stem Cell Biology. (1st edn). New York: Springer-Verlag.
- Dick JE (2005) Acute myeloid leukemia stem cells. Ann N Y Acad Sci 1044: 1-5. [Crossref]
- Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, et al. (2004) MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 6: 587-596. [Crossref]
- Warner JK, Wang JC, Hope KJ, Jin L, Dick JE (2004) Concepts of human leukemic development. Oncogene 23: 7164-7177. [Crossref]
- Adams GB, Chabner KT, Alley IR, Olson DP, Szczepiorkowski ZM, et al. (2006) Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 439: 599-603. [Crossref]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, et al. (2005) SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121: 1109-1121. [Crossref]
- Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, et al. (2005) Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 106: 1232-1239. [Crossref]
- Zhang J, Niu C, Ye L, Huang H, He X, et al. (2003) Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425: 836-841. [Crossref]
- Lymperi S, Horwood N, Marley S, Gordon MY, Cope AP, et al. (2008) Strontium can increase some osteoblasts without increasing hematopoietic stem cells. Blood 111: 1173-1181. [Crossref]
- Avecilla ST, Hattori K, Heissig B, Tejada R, Liao F, et al. (2004) Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med 10: 64-71. [Crossref]
- Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, et al. (2007) Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131: 324-336. [Crossref]
- Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, et al. (2007) Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 25: 1315-1321. [Crossref]
- Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, et al. (2008) Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell 13: 483-495. [Crossref]
- Al-Mawali A. (2013) Leukemic stem cells show the way for novel target of acute myeloid leukemia therapy. J Stem Cell Res Ther 3: 151.
- ten Cate B, de Bruyn M, Wei Y, Bremer E, Helfrich W (2010) Targeted elimination of leukemia stem cells; a new therapeutic approach in hemato-oncology. Curr Drug Targets 11: 95-110. [Crossref]
- Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, et al. (2005) In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435: 969-973. [Crossref]
- Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, et al. (2009) Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 113: 6215-6224. [Crossref]
- Doan PL, Chute JP (2012) The vascular niche: home for normal and malignant hematopoietic stem cells. Leukemia 26: 54-62. [Crossref]
- Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, et al. (2003) Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med 9: 1158-1165. [Crossref]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y (2004) Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 101: 7618-7623. [Crossref]
- Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, et al. (2009) Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 5: 31-42. [Crossref]
- Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE (2006) Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 12: 1167-1174. [Crossref]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, et al. (2010) The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 327: 1650-1653. [Crossref]
- Lutz C, Hoang VT, Buss E, Ho AD (2013) Identifying leukemia stem cells--is it feasible and does it matter? Cancer Lett 338: 10-14. [Crossref]
- Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, et al. (2010) Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464: 852-857. [Crossref]
- Walter RB, Appelbaum FR, Estey EH, Bernstein ID (2012) Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 119: 6198-6208. [Crossref]
- Annaloro C, Onida F, Saporiti G, Lambertenghi Deliliers G (2011) Cancer stem cells in hematological disorders: current and possible new therapeutic approaches. Curr Pharm Biotechnol 12: 217-225. [Crossref]
- Du X, Ho M, Pastan I (2007) New immunotoxins targeting CD123, a stem cell antigen on acute myeloid leukemia cells. J Immunother 30: 607-613. [Crossref]
- Frankel A, Liu JS, Rizzieri D, Hogge D (2008) Phase I clinical study of diphtheria toxin-interleukin 3 fusion protein in patients with acute myeloid leukemia and myelodysplasia. Leuk Lymphoma 49: 543-553. [Crossref]
- Sackstein R, Merzaban JS, Cain DW, Dagia NM, Spencer JA, et al. (2008) Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med 14: 181-187. [Crossref]
- Zhou J, Chng WJ1 (2014) Identification and targeting leukemia stem cells: The path to the cure for acute myeloid leukemia. World J Stem Cells 6: 473-484. [Crossref]
- Majeti R (2011) Monoclonal antibody therapy directed against human acute myeloid leukemia stem cells. Oncogene 30: 1009-1019. [Crossref]
- Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, et al. (2009) CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138: 271-285. [Crossref]
- Theocharides AP, Jin L, Cheng PY, Prasolava TK, Malko AV, et al. (2012) Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med 209: 1883-1899. [Crossref]
- Zhao X, Singh S, Pardoux C, Zhao J, Hsi ED, et al. (2010) Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica 95: 71-78. [Crossref]
- van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, et al. (2007) The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 110: 2659-2666. [Crossref]
- Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, et al. (2010) TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 7: 708-717. [Crossref]
- Kikushige Y, Miyamoto T (2013) TIM-3 as a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. Int J Hematol 98: 627-633. [Crossref]
- Gilliland DG, Jordan CT, Felix CA (2004) The molecular basis of leukemia. Hematology Am Soc Hematol Educ Program: 80-97. [Crossref]
- Piloto O, Nguyen B, Huso D, Kim KT, Li Y, et al. (2006) IMC-EB10, an anti-FLT3 monoclonal antibody, prolongs survival and reduces nonobese diabetic/severe combined immunodeficient engraftment of some acute lymphoblastic leukemia cell lines and primary leukemic samples. Cancer Res 66: 4843-4851. [Crossref]
- Knapper S, Burnett AK, Littlewood T, Kell WJ, Agrawal S, et al. (2006) A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood 108: 3262-3270. [Crossref]
- Karp J (2008) Future research directions for the treatment of AML. Clin Adv Hematol Oncol 6: 8-10. [Crossref]
- Linn YC, Hui KM (2010) Cytokine-induced NK-like T cells: from bench to bedside. J Biomed Biotechnol 2010: 435745. [Crossref]
- Maus MV, Grupp SA, Porter DL, June CH (2014) Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123: 2625-2635. [Crossref]