Abstract
Hepatic steatosis is the abnormal retention of lipids in hepatocytes, which can lead to progressive inflammation and fibrosis. Several dietary strategies including the use of low carbohydrate diets have been proposed to ameliorate the metabolic dysregulations caused by hepatic steatosis. Animal models are useful to study diet effects on hepatic lipid accumulation, inflammation and oxidative stress. Guinea pigs are an excellent animal model to study diet effects on cholesterol and lipoprotein metabolism, inflammation and atherosclerosis. The use of guinea pigs as models for hepatic steatosis has not been thoroughly discussed. The purpose of this review is to discuss the metabolic abnormalities that lead to the development of hepatic steatosis, to review some dietary treatments that can reverse these abnormalities and finally to propose the guinea pig as an animal model to study the metabolic pathways, signaling and genetic mechanisms associated with hepatic steatosis and non-alcoholic fatty liver disease.
Key words
guinea pigs, animal models, diet, hepatic steatosis, systemic inflammation
Abbreviations
4-HNE: 4-Hidroxy-2-nonenal; ALT: Alanine Aminotransferase; ATP: Adenosine Triphosphate; ER: Endoplasmic Reticulum; HDL-C: HDL-cholesterol; LDL-C: LDL-cholesterol; MAD: Malonaldehyde; NAD: Nicotinamide Dinucleotide; NAFLD: Non-alcoholic Fatty Liver Disease; NASH: Non-alcoholic Steatohepatis; PEPCK: Phosphoenol Pyruvate Carboxylase; PPAR-α: Peroxisome proliferator Activated Receptor-α; PUFA: Polyunsaturated Fatty Acids; ROS: reactive oxygen species; SFA: Saturated Fatty Acids; TC: Total Cholesterol; TG: Triglycerides; VLDL: Very Low Density Lipoprotein
Hepatic steatosis
Pathological conditions such as diabetes, insulin resistance, cardiovascular dysfunction, and non-alcoholic fatty liver disease (NAFLD), among others, are clear examples of distinct disturbances in metabolism both at cellular and systemic level [1]. Accelerated flux of metabolites (glucose, fructose, certain amino acids) through the lipogenic pathway, dysregulated synthesis of very low density lipoprotein (VLDL) particles [2], inefficient decarboxylation and oxidation of fatty acids (both mitochondrial and peroxisomal) [3], and inappropriate handling and storage of fatty acid pools [4] are the primary mechanisms at the level of the liver that are responsible for steatosis, the prerequisite of NAFLD. Other alterations external to the liver occur in the adipose tissue, where adipocytes are unable to respond to insulin’s suppressive effect on lipolysis, producing an increased mobilization of non-esterefied fatty acids to the liver [5]. The situation becomes even more daunting during the inflammatory transition of steatosis to non-alcoholic steatohepatitis (NASH) that could lead to advanced liver fibrosis, cirrhosis and hepatocellular carcinoma [6]. Metabolically stressed organelles including the mitochondria and endoplasmic reticulum (ER) react to a variety of cellular insults through pro-inflammatory mechanisms to produce the inflammatory infiltrates, ballooned hepatocytes and cross-linked fibrotic tracts that define NASH [7].
These metabolic interruptions within the liver extend throughout hepatocytes as well as stellate and Kupffer cells where cholesterol crystals can be localized [8]. To gain some insight as to why a broader, more encompassing metabolic perspective is appropriate in the definition of NAFLD, it is important to understand the lipocentric nature of the “two-hit” hypothesis [9]. Lipids, as fatty acids, are needed first to secure the development of steatosis, which emerges from an overload of the oxidizing systems responsible for fatty acid dysregulation. Such an overabundance of fatty acids increases their availability to serve as substrates for the principal microsomal cytochrome P450 enzymes, CYP2E1 and CYP4A. Normally these enzymes function is to hydroxylate non-polar substrates, including fatty acids, producing more polar compounds for eventual conjugation or excretion. However, as their catalytic activity is oxygen dependent, there are many harmful metabolites produced from this system and it is these superoxide anions (O2._), hydrogen peroxide (H2O2), and hydroxyl radicals (-OH), that when produced in amounts exceeding antioxidant species lead to a state of cellular oxidative stress [10]. Reactive oxygen species (ROS) can also be produced from complexes of the mitochondrial respiratory chain and because of their fairly short half-life, many of the cellular effects occur in a close proximity to mitochondria themselves; these local effects include the depletion of adenosine triphosphate (ATP) and nicotinamide dinucleotide (NAD), damage to cellular macromolecules such as DNA, protein denaturation, and not surprisingly the reduction of antioxidant defenses, in particular glutathione [11]. With limited capacity for cellular defense, free radicals (in particular the hydroxyl radical) are able to migrate unopposed into membrane lipid bilayers and attack vulnerable unsaturated side chains of membrane polyunsaturated fatty acids (PUFA). This in turn creates lipid hydroperoxides that release aldehyde bi-products, of which 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) are of considerable importance in biological systems [12]. Lipid peroxidation itself also acts as an amplifier of free radical and aldehyde generation, further depleting antioxidant capacity and thereby exacerbating the state of oxidative stress [13]. Lastly, aside from impairing organelle function, aldehydes such as 4-HNE and MDA can directly induce the production of cytokines, in particular tumor necrosis factor-α, which in turn acts as a chemo attractant for neutrophils leading to chemotaxis, inflammation, and eventually apoptotic-mediated cell death. Hepatic stellate cells, the principal mediators of liver fibrosis can also be directly activated following membrane lipid peroxidation through the production of transforming growth factor b [14,15].
Dietary strategies and hepatic steatosis
Several investigators have observed a strong association between excessive carbohydrate intake and an increased severity of hepatic steatosis and NASH in humans [16]. In a recent prospective study, Kang et al. [17] evaluated 91 patients with clinically suspected NAFLD and characterized 31 patients (34%) as having metabolic syndrome. Those patients with metabolic syndrome were shown to consume more carbohydrate (p =0.03) and less fat (p=0.01) and displayed higher histological severity scores for steatosis (p=0.004) and NASH (p =0.0006) compared to patients without metabolic syndrome. This particular study is noteworthy as it recapitulates current evidence regarding the link between excessive carbohydrate consumption and the development of physiological factors associated with metabolic syndrome as recently described [18]. A carbohydrate restricted dietary intervention was recently conducted in an obese female patient diagnosed with NAFLD [19] that presented abnormal plasma alanine amino transferase (ALT) levels and a hepatic triglyceride content of 44.6% as assessed by 1H MRS. The patient was instructed to follow for 2 weeks a ketogenic diet composed of 5: 41: 54 % carbohydrate: fat: protein respectively. At 5 weeks after the initiation of the diet, ALT values returned to normal and there was a remarkable reduction in hepatic triglyceride content to 11.9%. While long-term controlled human studies designed to evaluate the role of carbohydrate restriction on improvements in NAFLD are sparse, the available evidence does indeed support a beneficial role for this dietary intervention in ameliorating the associated clinical and histological abnormalities inherent in this disease. In an interesting animal study [20] two groups of mice were fed diets high in carbohydrate (68%) that differed in starch composition in order to examine the effects of rapidly absorbed carbohydrate on hepatic steatosis. One group consumed a diet composed of 100% amylopectin, a highly branched chain glucose polymer which is rapidly metabolized within the small intestine, whereas the other group consumed 60% amylose and 40% amylopectin; amylose due to its linear structure, is hydrolyzed at a slower rate and therefore predicted to contribute to less severe hepatic steatosis. Indeed, histological examination indicated the presence of mixed hepatic steatosis in mice fed the rapidly absorbed carbohydrate and a two-fold greater hepatic triglyceride content (20.7 + 9.4 mg/g vs. 9.6 + 4.9 mg/g; p=0.01) when compared to the mice that consumed the slower absorbed carbohydrate. Again, long term clinical studies which examine the relationship between carbohydrate and NAFLD in humans are warranted, however this study clearly points to the potential benefit(s) of reduced carbohydrate or low glycemic index diets as a novel dietary treatment for reversing the progression of hepatic steatosis.
To date, there have been relatively few long-term controlled studies investigating the role of dietary composition, in particular, carbohydrate and carbohydrate restriction on NAFLD although several lines of evidence suggest that limited carbohydrate consumption may be beneficial towards improved liver function. In a one year prospective study, Solga et al. [16] reported that a higher carbohydrate intake was associated with higher odds of hepatic inflammation whereas a higher fat intake was associated with lower odds of inflammation in morbidly obese patients with NAFLD. In addition, the authors observed a trend toward lower odds of steatosis and fibrosis in the highest tertile of fat intake suggesting that the presence of fat may improve NAFLD histopathology. The underlying mechanism(s) pertaining to carbohydrate-induced hepatic inflammation have yet to be clarified. A more recent study by Tendler et al. [21] found that a low carbohydrate, ketogenic diet (< 20 g/day carbohydrate) significantly improved steatosis, inflammation, and fibrosis in NAFLD patients within a 6 month period concomitant with weight loss and lowered plasma insulin and glucose levels. These results support the prevailing notion that insulin resistance and hyperglycemia play salient roles in enhancing hepatic DNL and triglyceride synthesis thus diets aimed to reduce insulin release may mitigate carbohydrate-stimulated lipogenesis.
Dietary fat, in particular polyunsatured fatty acids (PUFA) are known to exert hypotriglyceridemic effects and are a potentially important dietary treatment in reducing cardiovascular disease risk. Evidence is also accumulating that suggests these may also exert various hepatoprotective effects. These fatty acids are well known regulators of hepatic gene expression with regard to carbohydrate and lipid metabolism, suppressing L-PK in the glycolytic pathway as well as several key lipogenic enzymes [22]. Sterol regulatory element binding protein (SREBP)-1c is potently inhibited by PUFAs and although the mechanistic details require further studies, it has been demonstrated that PUFAs reduce SREBP-1c transcription, enhance SREBP-1c mRNA turnover and directly interfere with the proteolytic cleavage of the transcriptionally mature SREBP-1c protein [22]. It has also been recently demonstrated that PUFAs, specifically linoleate (18:2), eicosapentanoic acid (EPA) and docosahexaenoic acid (DHA) effectively inhibit ChREBP activity in mice, thereby decreasing expression of both L-PK and FAS [23]. A recent study investigating the potential therapeutic effects of omega-3 fatty acids in patients clinically diagnosed with hepatic steatosis [24] found that those receiving 2.7 grams daily of concentrated EPA for twelve months had significantly reduced plasma values of ALT and AST and a regression of steatosis as assessed by histological analysis. An eight week trial in patients with elevated hepatic TG content (as assessed by proton magnetic resonance spectroscopy) who received 9 grams/day of fish oil showed a 46% reduction in plasma TG, a 21% reduction in plasma VLDL and IDL cholesterol and a 15% decrease in plasma apolipoprotein B, yet no improvement in hepatic TG content [25]. It must be mentioned that the sample population for this study was fairly small (n=17) and included mainly African-Americans, who tend to accumulate less intraperitoneal fat and differ from other ethnic groups such as Hispanics and Caucasians in terms of the prevalence of steatosis [26].
Nutritional modulation of hepatic anomalies such as steatosis and the more severe NASH through carbohydrate restriction as well as increased unsaturated fatty acids are beginning to show promise as front-line dietary defenses. Although the evidence is still fairly limited for these approaches the metabolic basis seems fundamentally sound. Reduced dietary intake of carbohydrate, particularly rapidly absorbed sources such as refined grains and starches restricts insulin release and shifts metabolism towards a state of energy-deficit, prompting the oxidation of stored carbohydrate, as glycogen and fat, as adipose TG. Dietary unsaturated fatty acids appear to function at the molecular level to antagonize the transcriptional regulators responsible for inducing the lipogenic program. While drugs such as insulin sensitizers and nuclear receptor agonists are valuable in their own right, it is time for nutritional strategies to emerge as viable therapeutic modalities for diseases of metabolic origin. Given the strategic role of the liver in managing incoming dietary components, this seems a logical place to start.
Use of animal models
The range of animal models that are currently used for non-alcoholic fatty liver disease (NAFLD) research is extensive and involves an array of both genetic and dietary models, the majority of which are mice and rats [27,28]. Genetically altered models of hepatic steatosis often display increased hepatic lipogenesis as is the case of phosphoenol pyruvate carboxylase (PEPCK)-n-SREBP-1α mice or decreased hepatic fatty acid oxidation like the peroxisome proliferator activated receptor-α (PPARα) -/- mice. The ob/ob mouse and fa/fa rat have naturally occurring mutations in the synthesis of leptin, a satiety hormone produced predominantly by white adipocytes, and in the gene encoding the leptin receptor, respectively [29]. As a consequence these animals exhibit severe disturbances in energy metabolism and develop hyperphagia, hyperinsulinemia, hyperglycemia, dyslipidemia, and spontaneously fatty livers [29] thereby making them a worthy model for NAFLD etiology. Phenotypically normal rodents have also been utilized to investigate the effects of dietary composition and individual nutrients on hepatic steatosis [27]. Rodent diets high in fat, sucrose, or fructose have been developed to enhance hepatic lipogenesis while methionine- and/or choline deficient diets are often implemented to impair fat oxidation [30]. Taken together, these genetic and dietary rodent models constitute the conceptual framework for our current understanding of the molecular and metabolic processes germane to steatosis and NAFLD.
The reality however, is that mice and rats differ substantially from humans in the genetic expression and activity level of several genes involved in hepatic lipid and cholesterol metabolism, thus the pathogenesis of this disease in these animals does not appropriately emulate that of the human situation [31]. Rather than relying solely on the tools of molecular biology to procure transgenic or genetic knock-out animals researchers must also direct their efforts towards identifying species that naturally replicate human metabolism. An ideal model should exhibit similarities in the pathway(s) of interest and respond to dietary, exercise, or pharmacological treatments in a manner analogous to that of human interventions. The guinea pig is perhaps the most excellent representation of such a model with regards to hepatic and whole body trafficking and processing of lipids and cholesterol and may represent a novel template for elucidating the true mechanisms implicated in human steatosis and NAFLD [32].
The most drastic metabolic difference between guinea pigs and rodents is that in guinea pigs the majority of cholesterol is transported within LDL particles [33], whereas rats or mice predominantly carry cholesterol in HDL particles [33]. This observation has made guinea pigs the ultimate animal model for human lipoprotein metabolism. Furthermore, similar to humans, guinea pigs display a high LDL-to-HDL ratio [34] and possess several key enzymes involved in the intravascular processing of lipoproteins, notably CETP, lecithin cholesterol acyltransferase (LCAT), and liporprotein lipase (LPL) [35]. These inherent similarities between guinea pigs and humans implicate this animal as a bona fide model for lipoprotein metabolism yet may also expand current knowledge pertaining to NAFLD and cardiovascular disease. NAFLD has been proposed as the hepatic manifestation of the metabolic syndrome [36] and is also closely linked to cardiovascular dysfunction [37,38]. Given the striking similarities between humans and guinea pig lipoprotein metabolism, in conjunction with recent research that has suggested guinea pigs as a suitable model to evaluate inflammation and atherosclerosis [36,39] it thus appear that guinea pigs may represent an innovative approach to discerning the mechanisms by which NAFLD contributes to the pathophysiology of cardiovascular disease.
Guinea pigs also resemble humans in the synthesis, storage, and catabolism of cholesterol in the liver. This observation is particularly relevant for hepatic steatosis and NAFLD, as emerging research has suggested cholesterol as a central molecule in the transition from hepatic steatosis to NASH [40]. Although excessive cholesterol storage may not solely cause hepatic steatosis per se, the accumulation of free cholesterol may exacerbate the degree of steatosis and hepatocellular dysfunction in humans [41]. Both humans and guinea pigs exhibit higher concentrations of hepatic free cholesterol versus esterified cholesterol [35,42] as well as similar enzyme activities of ACAT, the ER-localized enzyme that esterifies cholesterol, and HMG-CoA reductase [43]. Moreover, guinea pigs demonstrate moderate rates of hepatic cholesterol synthesis [44] and catabolism [45] comparable to humans. While it remains unclear as to whether the expression and activity of enzymes involved in hepatic fatty acid synthesis and combustion correlate between guinea pigs and humans, the striking similarities in hepatic cholesterol metabolism warrant the use of this animal as a novel model for elucidating the role of cholesterol in the progression of hepatic steatosis and the transition to more advanced stages of NAFLD. Lastly, guinea pigs have been repeatedly demonstrated to respond to dietary interventions, including carbohydrate restriction [46], fatty acid composition [47] and dietary fiber [45] in a manner analogous to humans [48,49] and given the potential therapeutic role of carbohydrate restriction in attenuating NAFLD, guinea pigs may represent the most appropriate means to evaluate the role of dietary composition on NAFLD, thus standing at the crossroads between laboratory research and clinical applications. Figure 1 shows key similarities between guinea pigs and humans documenting their potential use as models for hepatic steatosis.
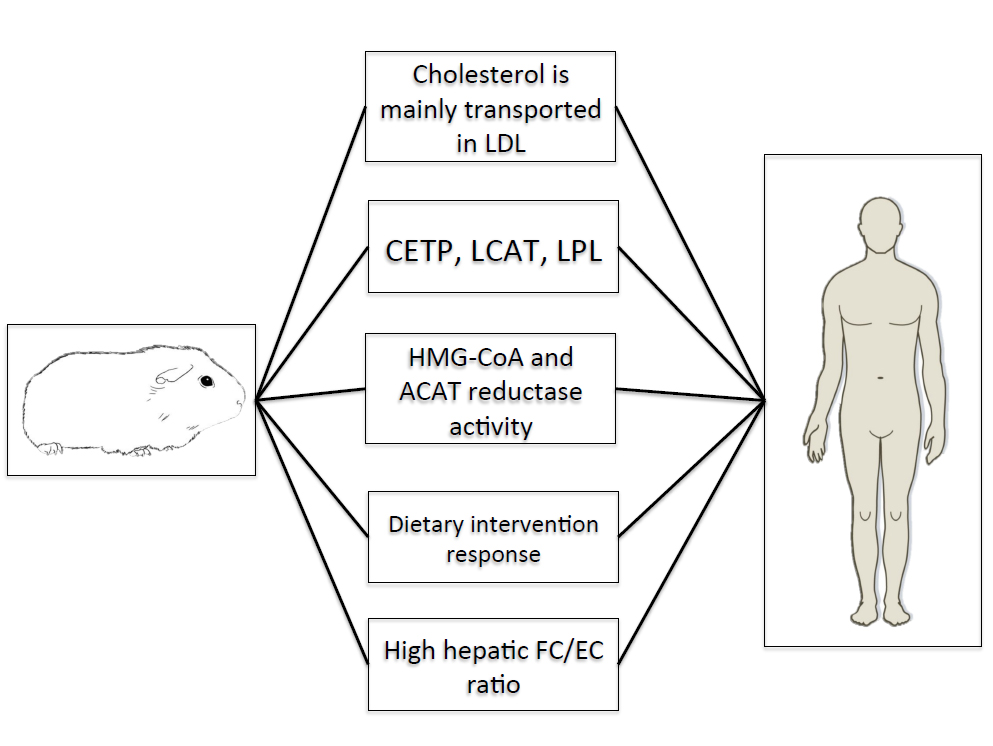
Figure 1. Key similarities between guinea pigs and humans
Conclusion
We conclude that guinea pigs can be used as reliable models for hepatic injury, hepatic steatosis and ultimately hepatic cirrhosis without undergoing any genetic manipulations. The fact that lipoprotein metabolism and that hepatic enzyme activity mimic the human situation strengthens further the possibility of using them as models to understand molecular mechanisms and alterations of metabolic pathways associated with hepatic steatosis.
References
- Rusu E, Enache G, Jinga M, Dragut R, Nan R, et al. (2015) Medical nutrition therapy in non-alcoholic fatty liver disease-a review of literature. J Med Life 8: 258-262. [Crossref]
- Fabbrini E, Magkos F (2015) Hepatic Steatosis as a Marker of Metabolic Dysfunction. Nutrients 7: 4995-5019. [Crossref]
- Auguet T, Berlanga A, Guiu-Jurado E, Martinez S, Porras JA, et al. (2014) Altered fatty acid metabolism-related gene expression in liver from morbidly obese women with non-alcoholic fatty liver disease. Int J Mol Sci 15: 22173-22187. [Crossref]
- Gusdon AM, Song KX, Qu S (2014) Nonalcoholic Fatty liver disease: pathogenesis and therapeutics from a mitochondria-centric perspective. Oxid Med Cell Longev 2014: 637027. [Crossref]
- Gentile CL (2015) The role of viscedral and subcutaneous adipose tissue fatty acid composition in liver pathophysiology associated with NAFLD. Adipocyte 4: 101-112.
- Krawczyk M, Bonfrate L, Portincasa P (2010) Nonalcoholic fatty liver disease. Best Pract Res Clin Gastroenterol 24: 695-708. [Crossref]
- Ikura Y (2014) Transitions of histopathologic criteria for diagnosis of nonalcoholic fatty liver disease during the last three decades. World J Hepatol 6: 894-900. [Crossref]
- Ioannou GM (2015) Cholesterol-lowering drugs cause distribution of cholesterol crystals and disperse Kupffer cell crown-like structures during resolution of NASH. J Lipid Res 56: 227-285.
- Day CP, James OF (1998) Steatohepatitis: a tale of two "hits"? Gastroenterology 114: 842-845. [Crossref]
- Robertson G (2001) Nonalcoholic steatosis and steatohepatitis: II. Cytochrome P-450 enzymes and oxidative stress. Am J. Physiol Gatrointes Liver Physiol 281: G1135-G1139.
- Angulo P (2002) Nonalcoholic fatty liver disease. N Engl J Med 346: 1221-1231. [Crossref]
- Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11: 81-128. [Crossref]
- Gardner HW (1989) Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic Biol Med 7: 65-86. [Crossref]
- Leonarduzzi G (1997) The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor ß-1 expression in the macrophage lineage: a link between oxidative injury and fibrosclerosis. FASEB J 11: 851-857.
- Uchida K (2003) 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res 42: 318-343. [Crossref]
- Solga S, Alkhuraishe AR, Clark JM, Torbenson M, Greenwald A, et al. (2004) Dietary composition and nonalcoholic fatty liver disease. Dig Dis Sci 49: 1578-1583. [Crossref]
- Kang H (2006) Metabolic syndrome is associated with greater histology severity, higher carbohydrate and lower fat diet in patients with NAFLD. Am. J. Gastroenterol 101: 2247-2253.
- Volek JS, Feinman RD (2005) Carbohydrate restriction improves the features of metabolic syndrome. Metabolic syndrome may be defined by the response to carbohydrate restriction. Nutr Metab (Lond) 2: 31.
2021 Copyright OAT. All rights reserv
- Browning JD (2006) A low-carbohydrate diet rapidly and dramatically reduces intrahepatic triglyceride content. Hepatology 44: 487-488.
- Scribner KB, Pawlak DB, Ludwig DS (2007) Hepatic steatosis and increased adiposity in mice consuming rapidly vs. slowly absorbed carbohydrate. Obesity (Silver Spring) 15: 2190-2199. [Crossref]
- Tendler D, Lin S, Yancy WS Jr, Mavropoulos J, Sylvestre P, et al. (2007) The effect of a low-carbohydrate, ketogenic diet on nonalcoholic fatty liver disease: a pilot study. Dig Dis Sci 52: 589-593. [Crossref]
- Jump DB (2008) N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Curr Opin Lipidol 19: 242-247. [Crossref]
- Denti R (2005) Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin Invest 115: 2843-2854.
- Tanaka N, Sano K, Horiuchi A, Tanaka E, Kiyosawa K, et al. (2008) Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J Clin Gastroenterol 42: 413-418. [Crossref]
- Vega GL, Chandalia M, Szczepaniak LS, Grundy SM (2008) Effects of N-3 fatty acids on hepatic triglyceride content in humans. J Investig Med 56: 780-785. [Crossref]
- Guerrero R, Vega GL, Grundy SM, Browning JD (2009) Ethnic differences in hepatic steatosis: an insulin resistance paradox? Hepatology 49: 791-801. [Crossref]
- Sanches SC, Ramalho LN, Augusto MJ, da Silva DM, Ramalho FS (2015) Nonalcoholic Steatohepatitis: A Search for Factual Animal Models. Biomed Res Int 2015: 574832. [Crossref]
- Koteish A, Mae Diehl A (2002) Animal models of steatohepatitis. Best Pract Res Clin Gastroenterol 16: 679-690. [Crossref]
- Koteish A, Diehl AM (2001) Animal models of steatosis. Semin Liver Dis 21: 89-104. [Crossref]
- Takai K, Funaba M, Matsui T (2016) Steatohepatitis is developed by a diet high in fat, sucrose, and cholesterol without increasing iron concentration in rat liver. Biol Trace Elem Res 170: 401-409. [Crossref]
- Fernandez ML (2001) Guinea pigs as models for cholesterol and lipoprotein metabolism. J Nutr 131: 10-20. [Crossref]
- deOgburn R (2015) Guinea pigs present hypercholesterolemia, hepatic steatosis and liver injury congruent with cholesterol-induced non-alcoholic fatty liver disease following a dietary cholesterol challenge. EC Nutrition 1: 28-40.
- Fernandez ML, McNamar DJ (1989) Dietary fat-mediated changes in hepatic apoprotein B/E receptor in the guinea pig: effect of polyunsaturated, monounsaturated, and saturated fat. Metabolism 38: 1094-1102. [Crossref]
- Sharman MJ (2008) Replacing dietary carbohydrate with protein and fat decreases the concentration of small LDL and the inflammatory response induced by atherogenic diets in the guinea pig. J Nutr. Biochem 19: 732-738.
- Fernandez ML, Conde AK, Ruiz LR, Montano C, Ebner J, et al. (1995) Carbohydrate type and amount alter intravascular processing and catabolism of plasma lipoproteins in guinea pigs. Lipids 30: 619-626. [Crossref]
- Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, et al. (2003) Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 37: 917-923. [Crossref]
- Malik S (2004) Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease and all causes in United States adults. Circulation 110: 1245-150.
- Villanova N, Moscatiello S, Ramilli S, Bugianesi E, Magalotti D, et al. (2005) Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 42: 473-480. [Crossref]
- Fernandez ML, Volek JS (2006) Guinea pigs: a suitable animal model to study lipoprotein metabolism, atherosclerosis and inflammation. Nutr Metab (Lond) 3: 17. [Crossref]
- Van Rooyen DM (2011) Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 141:1393-1403.e5
- Arguello G, Balboa E, Arrese M, Zanlungo S (2015) Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim Biophys Acta 1852: 1765-1778. [Crossref]
- Angelin B, Olivecrona H, Reihnér E, Rudling M, Ståhlberg D, et al. (1992) Hepatic cholesterol metabolism in estrogen-treated men. Gastroenterology 103: 1657-1663. [Crossref]
- Roy S, Vega-Lopez S, Fernandez ML (2000) Gender and hormonal status affect the hypolipidemic mechanisms of dietary soluble fiber in guinea pigs. J Nutr 130: 600-607. [Crossref]
- Fernandez ML, Yount NY, McNamara DJ (1990) Whole body and hepatic cholesterol synthesis rates in the guinea-pig: effect of dietary fat quality. Biochim Biophys Acta 1044: 340-348. [Crossref]
- Fernandez ML (1995) Distinct mechanisms of plasma LDL lowering by dietary fiber in the guinea pig: specific effects of pectin, guar gum, and psyllium. J Lipid Res 36: 2394-2404. [Crossref]
- deOgburn R (2012) Increased dietary cholesterol in conjunction with carbohydrate restriction uniquely impacts hepatic lipid metabolism in a guinea pig model. Comparative Medicine 62: 109-115.
- Torres-Gonzalez M, Shrestha S, Sharman M, Freake HC, Volek JS, et al. (2007) Carbohydrate restriction alters hepatic cholesterol metabolism in guinea pigs fed a hypercholesterolemic diet. J Nutr 137: 2219-2223. [Crossref]
- Torres-Gonzalez M (2008) Carbohydrate restriction and dietary cholesterol distinctly affect plasma lipids and lipoprotein subfractions in adult guinea pigs. J Nutr Biochem 19: 856-863.
- Leite JO, Vaishnav U, Puglisi M, Fraser H, Trias J, et al. (2009) A-002 (Varespladib), a phospholipase A2 inhibitor, reduces atherosclerosis in guinea pigs. BMC Cardiovasc Disord 9: 7. [Crossref]