Enzymes requiring adenosylcobalamin (AdoCbll) or complexes of [4Fe–4S]1+-S-adenosylmethionine (AdoMet) function with the intermediate formation of the 5’-deoxyadenosyl radical and catalyze reactions involving carbon-centered radical intermediates. The latter form the radical SAM superfamily and are far more numerous and catalyze much more chemically diverse reactions than the AdoCbl-enzymes. The radical SAM superfamily likely preceded the AdoCbl-enzymes in evolution.
As a graduate student under R. H. Abeles In 1964-1968, the author investigated the adenosylcob(III)alamin-dependent reaction of dioldehydrase (DDH), a bacterial enzyme catalyzing the dehydration of propane-1,2-diol to propionaldehyde according to equation 1.
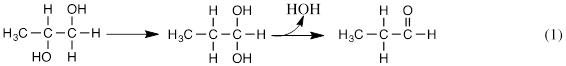
The reaction proceeded with internal transfer of a hydrogen from C1 to C2 and transfer of C2(OH) to C1 [1]. The mechanistic role of adenosylcob(III)alamin (AdoCbl), a Vitamin B12 coenzyme, was of interest. AdoCbl had been found to include a covalent bond between Co(III) of the vitamin and C5’ of the adenosyl group [2]. The cobalt—carbon bond was found to be weak and subject to homolytic scission, as illustrated in Figure 1, where 5’-dAdo• is the 5’-deoxyadenosyl radical.
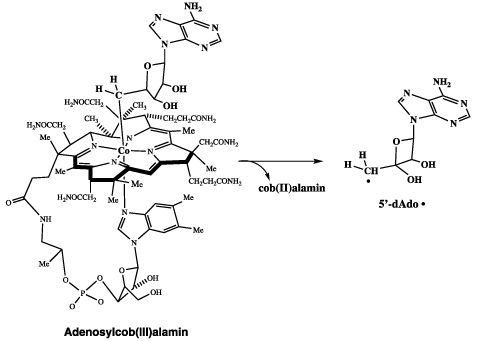
Figure 1. Homolytic cleavage of the Co-C5’ bond in adenosylcobalamin to 5’-dAdo . radical.
The substrate analogue glycolaldehyde had been shown to inactivate dioldehydrase in the presence of AdoCbl, with irreversible cleavage of the C—Co(III) bond to form cob(II)alamin [3]. The author undertook to determine the fate of the 5’-adenosyl group and proved it to be 5’-deoxyadenosine [3], so that the inactivation could be described as in equation 2 [4]. Inactivation of DDH by glycolaldehyde proved to be the first mechanism of suicide inactivation of an enzyme by a substrate analogue to be described in chemical terms.
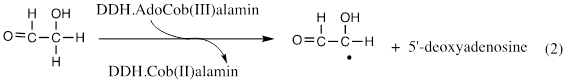
The author further showed that inactivation of DDH by [2-3H]glycolaldehyde led to [3H]5’-deoxyadenosine, suggesting that the 5’-deoxyadenosyl group in AdoCbl might participate in hydrogen transfer; and he proved this in the catalytic reaction by showing that the reaction of [1-3H]propane-1,2-diol as a substrate produced [5’-3H]AdoCbl at the active site of DDH [5,6]. Reaction of [5’-[3H]AdoCbl with DDH and propane-1,2-diol produced [3H]propionaldehyde.
The foregoing observations led to a reaction mechanism initiated by the 5’-deoxyadenosyl radical and proceeding at the active site of DDH through a sequence of carbon-centered radicals, as depicted in Figure 2.
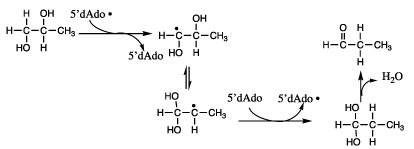
Figurge 2. The chemical roles of the 5’-deoxyadenosyl radical and other carbon-centered radicals at the active site of dioldehydraste (DDH).
In addition to DDH, reactions of other AdoCbl-dependent isomerases, including glutamate mutase, methylmalonyl-CoA mutase, glycerol dehydratase, lysine 5,6-aminomutase, and ornithine 4,5-aminomutase, follow the pattern of equation 3, β-interchange of hydrogen and another group between adjacent carbon atoms (equation 3). As subsequently shown, adenosyl-C5’ of AdoCbl mediates hydrogen transfer in each reaction [7].
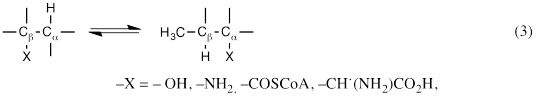
An exception is the AdoCbl -dependent ribonucleotide reductase [8,9], in which C5’ of AdoCbl initiates chemistry by hydrogen abstraction from Cys408 [10]. Thus, hydrogen transfer is proven for chemical reactions of all AdoCbl -dependent enzymes. Substrate-radical species, as detected in the reaction of DDH [11], are intermediates in all of these reactions [7].
Prior to the 1980s, AdoMet was known as the biological methylating agent, for its role in donating the methyl group to numerous substrates engaged in essential biological functions. These reactions proceeded with methyl transfer by AdoMet in a polar displacement mechanism to nucleophilic nitrogen or oxygen atoms in the substrates to form the methylated products and S -adenosylhomocysteine.
An exception to methyl transfer in the activation of pyruvate formate lyase (PFL) proved to be essential [12], in which AdoMet was cleaved by the PFL activase to 5’-deoxyadenosine [13], concomitant \with the production of a radical form of PFL, identified as a glycyl radical [14].
The author and his associates discovered the mechanistic role of AdoMet in the reaction of lysine 2,3-aminomutase (LAM), which had been discovered by H. A. Barker and his associates in 1970 and remained obscure in the literature until 1987 [15]. The enzyme catalyzed the interconversion of L-α-lysine and L-β-lysine according to the pattern of AdoCbl-dependent reactions (equation 3) but did not require a Vitamin B12 coenzyme. LAM required AdoMet and pyridoxal-5’-phosphate, and the activity was increased by the presence of added iron and a reducing agent. To determine whether the 5’-adenosyl group of AdoMet mediated hydrogen transfer, as in AdoCbl, the action of LAM on L-lysine was examined upon activation with S-[5’-3H]adenosylmethionine [16]. This reaction produced the equilibrium mixture of L-a-[3H]lysine and L-b-[3H]-lysine, proving hydrogen transfer by the 5’-deoxyadenosyl group of AdoMet. Deuterium transfer experiments proved that hydrogen transfer took place both inter-and intramolecularly [17], as required if 5’-deoxyadenosine is an intermediate in hydrogen transfer.
The author and his associates proposed that LAM somehow produced the 5’-deoxyadenosyl radical from AdoMet, and that this radical initiated the chemical mechanism in Figure 3. Of the four carbon-centered radicals in Figure 3, three have been observed and characterized spectroscopically and shown to be kinetically competent intermediates [18-20]. Radical 3 was the dominant radical in the steady state in the reaction of L-lysine [18]. 4-Thia-L-lysine proved to be a substrate, and the 4-thia-analog of radical 1 proved to be the dominant radical in the steady state [19]. An analogue of AdoMet, S-[3’,4’-anhydro]adenosylmethionine, proved to be fully functional, and the allylic analogue of 5’-dAdo • , 3’,4’-anhydro-5’-Ado •, was the dominant radical in the steady state [20].
To address the question of how the 5’-deoxyadenosyl radical (5’-Ado • in Figure 3) could arise from AdoMet, the author and associates investigated the role of iron in the action of LAM. They found that LAM, purified under strictly anaerobic conditions, contained a [4Fe–4S]1+ cluster that was required for activity [21,22]. Oxidation of the cluster to [3Fe–4S]2+ eliminated activity. Apparent cleavage of the strong S-C5’ bond in AdoMet (>60 kcal mol-1) required electron transfer, presumably from the [4Fe–4S]1+ cluster. Like the pyruvate formate lyase (PFL) activating enzyme [23], AdoMet was found to be ligated directly to Fe in the [4Fe4S] cluster, as illustrated in Figure 4 [24]. Intimate binding between the cluster and AdoMet facilitated electron transfer from {Fe–4S]1+ to AdoMet and cleavage to the 5’-deoxyadenosyl radical, as shown in Figure 4. This cleavage of AdoMet to the 5’-deoxyadenosyl radical consolidated the parallel between the functions of AdoCbl and AdoMet.
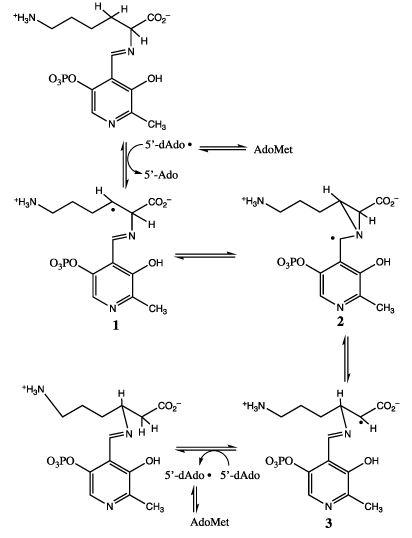
Figure 3. The carbon-centered radical mechanism in the interconversion of lysine and β-lysine by lysine 2,3-aminomutase.
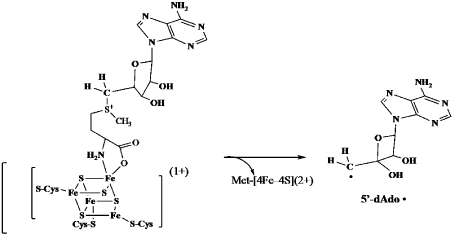
Figure 4. Homolytic cleavage of the S-C5’ bond in adenosylmethionine to 5’-dAdo . radical
In the 1990s several other enzymes not engaged in methyl transfer became known to require AdoMet and iron-sulfur clusters. These included anaerobic ribonucleotide reductase, biotin synthase and lipoyl synthase [25-33]. The latter two catalyzed the insertion of sulfur into unreactive C—H bonds of precursors. The ribonucleotide reductase in E. coli grown anaerobically and activated by AdoMet proved to contain a [4Fe–4S] cluster. And the PFL activating enzyme was found to contain iron-sulfur clusters, and when purified anaerobically to contain a [3Fe–4S] cluster [34,35]. All of the reactions of these enzymes were found to proceed by radical mechanisms.
The radical SAM superfamily
In due course the nucleotide sequences of genes encoding the foregoing AdoMet /[4Fe–4S]-dependent enzymes became known. Heidi L. Sofia and her associates examined the translated amino acid sequences and discovered the motif CxxxCxxC in common among these enzymes. They searched the available genomic databases in 2001 and found nearly 600 homologs, which they named the Radial SAM superfamily [36]. They found superfamily members throughout biology in both the animal and plant kingdoms. With the passage of time, the genomic database grew to include more than 100,000 homologous sequences in the Radical SAM superfamily encoding enzymes engaged in more than 70 amazingly diverse functions [37-39].
Members of the superfamily include enzymes that catalyze nucleic acid and protein modifications, enzymes that catalyze key steps in biosynthesis of vitamins, coenzymes, analogues of nucleotide bases, antibiotics and many other biological species. A number of Radical SAM enzymes require both AdoCbl and AdoMet and catalyze methylations of non-nucleophilic carbon and phosphorus atoms. Enzymes of the radical SAM superfamily might catalyze the most chemically diverse reactions of any superfamily in biochemistry.
Fewer than a third of all Radical SAM enzymes have been experimentally investigated in detail. Most of them appear to function by mechanisms involving carbon-centered radicals. However, in some of them, AdoMet displays bimodal functions, polar methylation in one step and aa a source of the 5’-deoxyadenosyl radical in another step [40,41]. It remains possible that one or another Radical SAM enzyme might engage only in polar methyl group transfer. Whether all Radical SAM enzymes catalyze carbon-centered radical chemistry remains to be determined in future research.
Origins
Cob(III)alamins are structurally complex, requiring many enzymatic steps to assemble. Adenosylcob(III)alamin synthetase produces AdoCbl in a single step from ATP and cob(I)alamin. Cobalamins are produced in bacteria as vitamin B12, and both bacteria and animals contain the synthetase. In contrast, de novo synthesis of the simpler methionine occurs only in plants, and it is a nutritionally essential amino acid for animals. iron-sulfrur clusters are among the most primitive cofactors. All things considered, the Radical SAM enzymes likely preceded AdoCbl enzymes in evolution.
The diverse radical SAM enzymes catalyze amino acid isomerizations; methylation of non-nucleophilic phosphorus [39] and carbon atoms [40,41]; modifications of proteins and nucleic acids, including deazaguanine biosynthesis [40]; glycyl radical formation; thiyl radical formation; chemically difficult steps in vitamin, cofactor, and antibiotic biosyntheses; and many other processes [37,38]. The less diverse AdoCbl proteins catalyze isomerizations and ribonucleotide reduction, and also servie as photosensors [43].
While a significant number of radical SAM enzymes have been found to function by way of the transient 5'-deoxyadenosyl radical, this is not a universal property of radical SAM enzymes. At least one member of the superfamily, the TsrM methylase, might not catalyze a radical reaction [44]. Another member of the superfamily catalyzes both radical and polar methylation [43]. Other members of the superfamily remain to be studied in detail.
LAM and its relative glutamate 2,3-aminomutase are as active or more active than AdoCbl dependent enzymes catalyzing analogous reactions. Therefore, the two coenzymatic systems should be equally efficient sources of the 5’-deoxyadenosyl radical. The sheer size of the radical SAM superfamily means that it is an evolutionary survivor, and AdoCbl enzymes are supplemental and mechanistically related sources of the 5'-deoxyadenosyl radical. In a possibly significant mechanistic advantage, AdoCbl produces the 5'-deoxyadenosyl radical without a requirement for a reducing agent. Radical SAM enzymes require a reducing system to generate the [4Fe–4S]1+ cluster in the cleavage of AdoMet (Figure 4).
The author is grateful to the many graduate and postdoctoral students who collaborated in the research described herein. Each and every participant made important contributions to this field. He is also grateful for the generous collaboration of his colleague Professor George H. Reed in guiding the spectroscopic characterization of carbon-centered radical intermediates. The author’s research in the radical SAM field was generously supported by the National Institute of Diabetes and Digestive and Kidney diseases of the USPHS.
- Abeles RH, Zagalak B (1966) The nature of the hydrogen transfer in the dimethylbenzimidazolylcobamide coenzyme-catalyzed conversion of 1,2-propanediol to propionaldehyde. J Biol Chem 241: 1245-1246. [Crossref]
- Lenhert PG, Hodgkin DC (1961) Structure of the 5,6-dimethyl-benzimidazolylcobamide coenzyme. Nature 192: 937-938. [Crossref]
- Wagner OW, Lee HA Jr., Frey PA, Abeles RH (1966) Studies on the mechanism of action of cobamide coenzymes. Chemical properties of the enzyme coenzyme complex. J Biol Chem 241: 1761-1762. [Crossref]
- Abend A, Bandarian V, Reed GH, Frey PA (2000) Identification of cis-ethanesemidione as the organic radical derived from glycolaldehyde in the suicide inactivation of dioldehydrase and of ethanolamine ammonia-lyase. Biochemistry 39: 6250-6257.
- Frey PA, Abeles RH (1966) The role of the B12 coenzyme in the conversion of 1,2-propanediol to propionaldehyde. J Biol Chem 241: 2732-2733. [Crossref]
- Frey PA, Essenberg MK, Abeles RH (1967) Studies on the mechanism of hydrogen transfer in the cobamide coenzyme-dependent dioldehydrase reaction. J Biol Chem 242: 5369-5377. [Crossref]
- Frey PA (2010) Cobalamin coenzymes in enzymology. In: Mander L, Liu, H-W, editors. Comprehensive Natural Products II Chemistry and Biology. Elsevier: Oxford 7: 501-546.
- Beck WS, Abeles RH, Robinson WG (1966) Transfer of hydrogen from cobamide coenzyme to water during ribonucleotide reduction. Biochem Biophys Res Commun 25: 421-425. [Crossref]
- Hogenkamp HP, Ghambeer RK, Brownson C, Blakely RL (1967) The relation of hydrogen exchange between deoxyadenosylcobalamin and water to the role of the coenzyme in ribonucleotide reduction. Biochem J 103: 5-7. [Crossref]
- Licht S, Gerfen GJ, Stubbe J (1996) Thiyl radicals in ribonucleotide reductases. Science 271: 477-481. [Crossref]
- Schepler KL, Dunham WR, Sands RH, Abeles RH (1975) Physical explanation of the EPR spectrum observed during catalysis by enzymes utilizing coenzyme B12. Biochim Biophys Acta 397: 510-518. [Crossref]
- Knappe J, Schmitt T (1978) A novel reaction of S-adenosyl-L-methionine correlated with the activation of pyruvate formate lyase. Biochem Biophys Res Commun 71: 1110-1117. [Crossref]
- Knappe J, Neugebauer FA, Blaschkowski HP, Gänzler M (1984) Post-translational activation introduces a free radical into pyruvate formate-lyase. Proc Natl Acad Sci USA 81: 1332-1335. [Crossref]
- Wagner AF, Frey M, Neugebauer FA, Schäfer W, Knappe J (1992) The free radical in pyruvate formate-lyase is located on glycine-734. Proc Natl Acad Sci U S A 89: 996-1000. [Crossref]
- Chirpich TP, Zappia V, Costilow RN, Barker HA (1970) Lysine 2,3-aminomutase. Purification and properties of a pyridoxal phosphate and S-adenosylmethionine-activated enzyme. J Biol Chem 245: 1778-1789. [Crossref]
- Moss M, Frey PA (1987) The role of S-adenosylmethionine in the lysine 2,3-aminomutase reaction. J Biol Chem 262: 14859-14862. [Crossref]
- Baraniak J, Moss ML, Frey PA (1989) Lysine 2,3-aminomutase. Support for a mechanism of hydrogen transfer involving S-adenosylmethionine. J Biol Chem 264: 1357-1360. [Crossref]
- Ballinger MD, Frey PA, Reed GH (1992) Structure of a substrate radical intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry 31: 10782-10789. [Crossref]
- Wu W, Lieder KW, Reed GH, Frey PA (1995) Observation of a second substrate radical intermediate in the reaction of lysine 2,3-aminomutase: A radical centered on the?-carbon of the alternative substrate, 4-Thia-L-lysine. Biochemistry 34: 10532-10537. [Crossref]
- Magnusson OT, Reed GH, Frey PA (2001) Charac2021 Copyright OAT. All rights reservthe 5'-deoxyadenosyl radical: an intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry 40: 7773-7782. [Crossref]
- Petrovich RM, Ruzicka FJ, Reed GH, Frey PA (1991) Metal cofactors of lysine 2,3-aminomutase. J Biol Chem 266: 7656-7660. [Crossref]
- Petrovich RM, Ruzicka FJ, Reed GH, Frey PA (1992) Characterization of iron-sulfur clusters in lysine 2,3-aminomutase by electron paramagnetic resonance spectroscopy. Biochemistry 31: 10774-10781. [Crossref]
- Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM (2002) An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron sited of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc 124: 11270-11271. [Crossref]
- Chen D, Walsby C, Hoffman BM, Frey PA (2003) Coordination and mechanism of reversible cleavage of S-adenosylmethionine by the [4Fe-4S] center in lysine 2,3-aminomutase. J Am Chem Soc 125: 11788-11789. [Crossref]
- Florentin D, Bui BT, Marquet A, Ohshiro T, Izumi Y (1994) On the mechanism of biotin synthase of Bacillus sphaericus. C R Acad Sci III 317: 485-488. [Crossref]
- Sanyal I, Cohen G, Flint DH (1994) Biotin synthase: purification, characterization as a [2Fe-2S] cluster protein, and in vitro activity of the Escherichia coli bioB gene product. Biochemistry 33: 3625-3631. [Crossref]
- Duin EC, Lafferty ME, Crouse BR, Allen RM, Sanyal I, et al. (1997) [2Fe-2S] to [4Fe-4S] cluster conversion in Escherichia coli biotin synthase. Biochemistry 36: 11811-11820. [Crossref]
- Hayden MA, Huang IY, Iliopoulos G, Orozco M, Ashley GW (1993) Biosynthesis of lipoic acid: characterization of the lipoic acid auxotrophs Escherichia coli W1485-lip2 and JRG33-lip9. Biochemistry 32: 3778-3782. [Crossref]
- Reed KE, Cronan JE Jr (1993) Lipoic acid metabolism in Escherichia coli: sequencing and functional characterization of the lipA and lipB genes. J Bacteriol 175: 1325-1336. [Crossref]
- Eliasson R, Fontecave M, Jornvall H, Krook M, Pontis E, Reichard P (1990) The anaerobic ribonucleoside triphosphate reductase from Escherichia coli requires S-adenosylmethionine as a cofactor. Proc Natl Acad Sci U S A 87: 3314-3318. [Crossref]
- Eliasson R, Pontis E, Fontecave M, Gerez C, Harder J, et al. (1992) Characterization of components of the anaerobic ribonucleotide reductase system from Escherichia coli. J Biol Chem 267: 25541-25547. [Crossref]
- King DS, Reichard P (1995) Mass spectrometric determination of the radical scission site in the anaerobic ribonucleotide reductase of Escherichia coli. Biochem Biophys Res Commun 206: 731-735. [Crossref]
- Mulliez E, Fontecave M, Gaillard J, Reichard P (1993) An iron-sulfur center and a free radical in the active anaerobic ribonucleotide reductase of Escherichia coli. J Biol Chem 268: 2296-2299. [Crossref]
- Broderick JB, Duderstadt RE, Fernandez DC, Wojtuszewski K, Henshaw TF, et al. (1997) Pyruvate formate-lyase activating enzyme is an iron-sulfur protein. J Am Chem Soc 119: 7396-7397.
- Broderick JB, Henshaw TF, Cheek J, Wojtuszewski K, Smith SR, et al. (2000) Pyruvate formate-lyase-activating enzyme: strictly anaerobic isolation yields active enzyme containing a [3Fe-4S](+) cluster. Biochem Biophys Res Commun 269: 451-456. [Crossref]
- Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, MillerNE (2001) Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional charctrization using new analysis and information visualization methods. Nucleic Acids Res 29: 1097-1106. [Crossref]
- Frey PA, Hegeman AD, Ruzicka FJ (2008) The Radical SAM Superfamily. Crit Rev Biochem Mol Biol 43: 63-88. [Crossref]
- Broderick JB, Duffus BR, Duschene KS, Shepard EM (2014) Radical S-adenosylmethionine enzymes. Chem Rev 114: 4229-4317. [Crossref]
- Hu K, Werner WJ, Allen KD, Wang SC (2015) Investigation of enzymatic C P bond formation using multiple quantum HCP nuclear magnetic resonance spectroscopy. Magn Res Chem 53: 267-272. [Crossref]
- McCusker KP, Medzihradszky KF, Shiver AL, Nichols RJ, Yan F, et al. (2012) Covalent intermediate in the catalytic mechanism of the radical S-adenosyl-L-methionine methyl synthase RlmN trapped by mutagenesis. J Am Chem Soc 134: 18074-18081. [Crossref]
- Schwalm EL, Grove TL, Booker SJ, Boal AK (2016) Crystallographic capture of a radical S-adenosylmethionine enzyme in the act of modifying tRNA. Science 352: 309-312. [Crossref]
- Bruender NA, Young AP, Bandarian V (2015) Chemical and Biological Reduction of the Radical SAM Enzyme 7-Carboxy-7-deazaguanine [corrected] Synthase. Biochemistry 54: 2903-2910. [Crossref]
- Padmanabhan S, Jost M, Drennan CL, et al. (2017) A New Facet of Vitamin B12: Gene Regulation by Cobalamin-Based Photoreceptors. Annu Rev Biochem 86: 485-514. [Crossref]
- Blaszzcyk AJ, Wang B, Silakov A, Ho JV, Booker SJ (2017) Efficient methylation of C2 in l-tryptophan by the cobalamin-depentdent radical S-adenosylmethionine methylase TsrM requires and unmodified N1 amine. J Biol Chem 292: 15456-15467. [Crossref]