Abstract
Primitive Neuroectodermal Tumor (PNET) is an aggressive round cell malignant tumor which belongs to Ewing’s Sarcoma family. Peripheral PNET is rare in head and neck region and even rare in palate with handful of cases reported till date. A case of Peripheral PNET of palate in a 32-year-old female patient along with its clinico-pathologic, radiologic and immunohistologic features as well as its management is reported here. We emphasize the need of histopathology and immunohistochemistry (IHC) for early diagnosis of such aggressive tumors to improve the prognosis of patient by providing timely management.
Key words
immunohistochemistry, palate, primitive neuroectodermal tumor (pnet), small round cell tumor
Introduction
A neuroectodermal tumor is a tumor of the central or peripheral nervous system. They are divided into two categories viz. Group (I) tumors, such as the pituitary adenomas and carcinoid tumors, represent tumors that show predominantly epithelial differentiation. Group (II) tumors, which incorporate malignant melanoma, olfactory neuroblastoma, Ewing’s sarcoma (EWS) and primitive neuroectodermal tumor that are predominantly neural and non-epithelial in origin [1].
Primitive neuroectodermal tumor (PNET) was first described by Hart and Earle in 1973 [2]. It is a small round cell tumor of presumed neural crest origin arising outside the central and sympathetic nervous system with variable differentiation [3]. Though primitive neuroectodermal tumors occur predominantly in central nervous system, peripheral PNETs have also been reported [4]. Peripheral PNETs are commonly seen in chest wall, trunks, abdomen and extremities of children and are relatively uncommon in head and neck region [5]. These tumors are aggressive and have a propensity for local recurrence and metastasis especially in bone marrow, brain, lungs and lymph nodes, but rarely to jaws and skull [1]. Primary peripheral PNET of maxilla is an extremely rare occurrence. The majority of the tumors are localized in the mandibular symphysis region and only ten cases are reported in the maxilla until now [6]. Recently, the incidence of PNETs in head and neck areas have been increasingly reported, however, there is a paucity of reports on PNETs of the oral cavity and that too in palate.
PNET affects primarily white and Hispanic adolescent or young individuals especially in the second decade of life with a male predominance and is a rare occurrence in people of African or Asian origin [6]. Our case is of thirty-two-year-old female of Indian origin. So, the age, gender and ethnicity of the patient in this case are unusual for Peripheral PNETs.
Emphasizing on the rarity of the lesion, a case of peripheral PNET of palate in an adult female patient along with its clinico-pathologic, radiologic, immunohistologic features and management is reported here.
Case report
A thirty-two years old female patient of Indian origin reported to the Out-Patient Department of our institute with a complaint of painful ulcer on her palate since five months. The ulcer was gradually progressive but has now suddenly increased in size since few days. It was associated with dull aching, intermittent and localized pain which aggravated on mastication and relieved with time. The medical and family histories were not of much significance.
Intraoral examination revealed a single ulcerative reddish lesion of size 3x2 cm approximately over left posterior region of palate extending antero-posteriorly from first premolar to second molar region and mesio-distally from midline to 1cm below the gingival margin of respective teeth (Figure 1). On palpation it was tender, with everted edges and indurated base. Provisional diagnosis of malignancy of palate was made with differential diagnosis of salivary gland malignancy and epithelial malignancy.
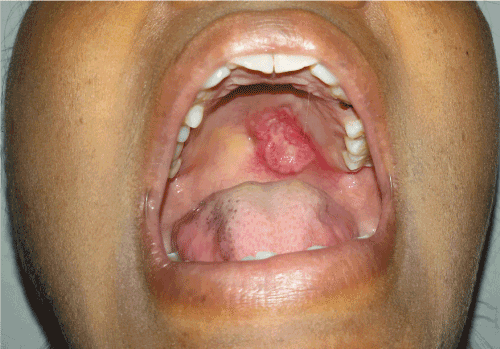
Figure 1. A single ulcerative lesion over left posterior region of palate
Computed Tomography (CT) showed bone resorption in bilateral palatine process of maxillary bone s/o malignant tumor or pressure resorption. Ultrasonography of neck revealed subcentrimetric lymphadenopathy in submental and bilateral submandibular region. On further examination, patient was found to be systemically fit with all the blood count values to be in normal range.
Incisional biopsy was performed under local anesthesia and the specimen was subjected to histopathological assessment. Under microscopic examination, low power view showed sheets of small to large lobules of discrete round cells. At places, round cells were arranged in the form of nests, strands and cords (Figure 2A). Surrounding connective tissue stroma was fibrocellular with myxoid areas at places; necrosis was evident (Figure 2B). Under higher magnification, mixed population of round cells was seen showing large round vesicular nuclei with rimming of eosinophilic cytoplasm and few with hyperchromatic nuclei. Few cells showed clear cytoplasm (Figure 2C). Nuclear pleomorphism was evident. At places 8-10 round cells were clustered in a circular fashion forming a vesicle in the centre suggestive of pseudorossettes (Figure 2D).
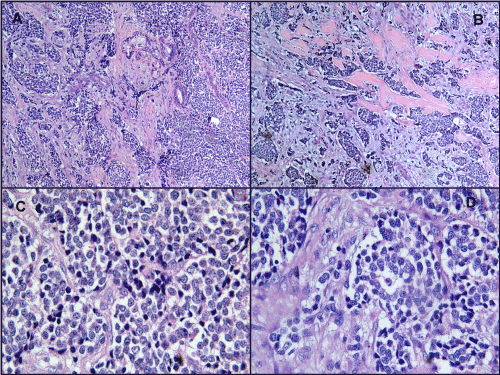
Figure 2A. Round cell arranged in the form of nests, strands and cords (100x H&E)
Figure 2B. Necrosis is seen as hyalinized eosinophilic areas (100x H&E)
Figure 2C. Mixed population of round cells showing large round vesicular nuclei with rimming of eosinophilic cytoplasm, few with hyperchromatic nuclei and few cells showing clear cytoplasm. Nuclear pleomorphism is seen (400x H&E)
Figure 2D. Pseudorossette (400x H&E)
Round cells can be seen in Ewing’s sarcoma family of tumors, rhabdomyosarcoma, osteosarcoma and lymphoma. To rule out Ewing’s sarcoma, Periodic Acid Schiff (PAS) stain was performed which came out negative. Immunhistochemistry (IHC) was performed to rule out another small round cell tumor which is tabulated in table 1. MIC-2 (CD99) (Figure 3A), Synapthophysin, Chromogranin A and Cytokeratin (Figure 3B) showed positive expressions and histopathologically pseudorossettes were seen, so a final diagnosis of PNET was given. On Ki67 immunostaining (Figure 3C), the tumor had a labeling index of 25-30% indicating moderate to high proliferation rate.
Table 1. IHC Markers for small round cell tumors
Marker |
Tumors |
Present case |
MIC-2 (CD99) |
PNET, Ewing’s sarcoma, Neuroendocrine carcinoma |
Positive |
Synaptophysin |
PNET, Ewings sarcoma, Neuroendocrine carcinoma |
Positive |
Chromagranin A |
PNET, Neuroendocrine carcinoma |
Positive |
Cytokeratin |
Neuroendocrine carcinoma, Rarely positive in PNET and Rhabdomyosarcoma. |
Positive |
Desmin |
Rhabdomyosarcoma |
Negative |
EMA (Epithelial membrane antigen) |
Lymphoma |
Negative |
LCA (Leukocyte common antigen) |
Lymphoma |
Negative |
TTF-1 (Thyroid transcription factor) |
Neuroendocrine carcinoma |
Negative |

Figure 3A. Round cells showing positive expression for CD99 (400x IHC)
Figure 3B. Round cells showing positive expression for Cytokeratin (400x IHC)
Figure 3C. Ki67 immunostaining (400x IHC)
Patient was then subjected to surgery under general anesthesia wherein wide local excision was done with more than 2 cm of clearance margin. Buccal fat pad was used as a reconstruction modality over other regional and distant flap as we needed a direct access to visualize the surgical site during follow up, as the recurrence rate of such tumors are on a higher side. Surgical obturator was provided to the patient for achieving proper functions and hygiene (Figures 4A and 4B). The resected specimen was then subjected to histopathological evaluation which confirmed the preoperative diagnosis.
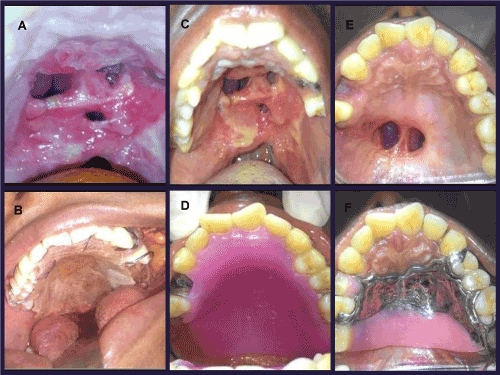
Figures 4A & 4B. Post-surgical oronasal fistula and surgical obturator
Figures 4C & 4D. Reduced size of oronasal fistula and interim obturator after three months
Figures 4E & 4F. Oronasal fistula after six months and definitive obturator
Patient is on close follow up and she is disease free and doing well. After three months interim obturator (Figures 4C and 4D) was given and it has been replaced with definitive obturator (Figures 4E and 4F) after six months of surgery. Fistula repair can be done in near future for the patient.
Discussion
Neuroectodermal tumors occupy a predominant place in oncology. Tumors exhibiting neuroectodermal differentiation occur throughout the body, and the diverse tissues of the head and neck give rise to a wide assortment of these neoplasms [7]. PNET is a term used to describe a category of neoplasm of neuroectodermal origin with variable cell differentiation [1]. The PNET are grouped as central and peripheral tumors. Central PNET such as medulloblastoma originates in the brain and spinal cord, whereas peripheral PNETs include adrenal and extra-adrenal neuroblastomas of soft tissues, nerve, and bone. The peripheral Primitive Neuroectodermal Tumor was first recognized by Arthur Purdy Stout in 1918 as a member of the family of “small round-cell tumors” which was subsequently designated as PNET [8].
Our case is of a thirty-two-year-old Indian female which is rare among the age group and ethnic distribution of tumor. PNET is seen involving any bone, however it predominately involves central and axial skeleton as well as the diaphysis of long bones and is unusual in head and neck region. Among head and neck region, it is infrequent in oral cavity with not many cases reported in maxilla. Considering all this rarity, this is an exceptional case.
Approximately 9% of PNET arise in the upper aerodigestive tract or head and neck region [9]. Stout reviewed twenty-six cases of PNET of head and neck region of which eleven tumors were localized to the mandible. Jurgens et al. carried out a retrospective analysis of forty-two cases, of which only four were localized to the head and neck region [10]. The lesions have an insidious onset, leading to large sizes at presentation. Distant metastases are identified in 20 to 25% of patients and are the adverse prognostic factors, especially in bone marrow, brain, lungs and rarely to lymph nodes [11].
The usual clinical manifestations include pain and swelling, however, it depends on the site of involvement. Intraoral PNET resembles commonly seen oral lesions such as, pyogenic granuloma; peripheral giant cell granuloma, lipoma and hemangioma. Minor salivary gland tumors can also resemble to soft tissue tumors [11]. Our case presented with painful reddish ulcer on palate which gave clinical appearance of salivary gland malignancy or epithelial malignancy.
The definitive diagnosis of PNET is based on the histological findings and IHC. PNET comes under a group of neoplasm which is collectively referred to as “small round cell tumors of childhood”, which include neuroblastomas, rhabdomyosarcoma, lymphoma, small round cell osteosarcoma and the Ewing family of tumors. Histopathologically, these neoplasms consist of sheets of poorly differentiated round cells with uniform, round to oval vesicular to hyperchromatic nuclei and scanty cytoplasm. However, it is not easy to distinguish PNET morphologically from other small round cell tumors and hence IHC is necessary for confirmation of the diagnosis. Discrimination criteria have been proposed to aid in diagnosis of PNET. Features are (1) well defined histological evidence of Homer-Wright or Flexner-Wintersteiner rosettes (Pseudorosettes). (2) Immunoreactivity to two or more neural markers, and/ or (3) ultra-structural transmission electron micrographic evidence of neural differentiation and neurosecretory granules [12]. We performed PAS stain which was negative, ruling out Ewings sarcoma. Osteosarcoma closely resembles to PNET but lack of osteoid tissue and presence of pseudorosettes ruled it out. Rhabdomyosarcomas are positive for desmin which was negative in our case. Lymphomas are positive for LCA which expressed negatively in this case. More than 90% of PNET will label strongly with antibodies directed against the MIC-2 protein product created by the t(11:22) (q24;q12) fusion of the EWS/FLI-1 genes and translocation t(21:22) (q22;q12) fusion of the EWS/ERG genes [13]. [[1]] This marker is of considerable diagnostic value. The positivity for CD99 (MIC-2), neural markers like synaptophysin and chromogranin A which are specifically stained in PNET and presence of pseudorossettes histopathologically confirmed the diagnosis of PNET. Cytokeratin came out positive in our case which is seldom positive in PNETs. Cytokeratin expression is present in about 20% of EWS/PNET in either diffuse or focal pattern [14]. Cytokeratin-positive PNET's have evidence of epithelial differentiation ultrastructurally, and may possibly represent a more aggressive subset of the EWS/PNET group of tumours [14]. Also, a 25-30% labeling index of ki67 immunostaining suggests high proliferative activity of tumor indicating poor prognosis.
With the advanced technology and research in the field of diagnostic and therapeutic oncology over the last few decades, identification of new and more effective treatment strategies and improved prognosis have been achieved. Local Surgical removal of the primary lesion along with combination of systemic chemotherapy to control subclinical micro-metastases should be considered for the treatment of PNET. The recommended treatment consists of a combination of chemotherapy, surgery and radiation therapy. Although PNET is considered radiosensitive, radiation is generally administered as an adjuvant therapy in cases of incomplete surgical re-section or where there are close tumor margins [12].
PNET has a tendency for recurrence and metastasis. Metastasis has been observed at the time of diagnosis in up to 31% of patients, reflecting the aggressiveness of these tumors. Metastasis to oral cavity from the primary lesion located at other sites is uncommon [1]. The prognosis of PNET is poor; with two-year survival rate in only 65% cases, after diagnosis, regardless of treatment modalities applied [11].
Conclusion
PNET itself is a rare entity in oral cavity; also, the location of tumor, age and ethnicity of patient is uncommon in this case. Also, Cytokeratin positivity and high proliferative rate of tumor cells indicate the aggressive nature of tumor in this case. Clinically it may suggest as minor salivary gland tumour or squamous cell carcinoma and therefore a clinician should always be aware of rare possibilities making histopathology and IHC the gold standard diagnostic modality. Early histopathologic diagnosis with timely management plays a crucial role in improving the prognosis of patient.
2021 Copyright OAT. All rights reserv
References
- Yuwanati M, Tupkari J, Mhaske S, Joshi P (2013) Primitive neuroectodermal tumor of the posterior mandible: A case report. J Clin Exp Invest 4: 101-104.
- Hart MN, Earle KM (1973) Primitive neuroectodermal tumors of the brain in children. Cancer 32: 890-897. [Crossref]
- Kadagad P, Umarani M (2011) Primitive Neuroectodermal Tumor of Maxilla: A Rare Case. WJD 2: 281-283.
- Dehner LP (1986) Peripheral and central primitive neuroectodermal tumors: A nosologic concept seeking a consensus. Arch Pathol Lab Med 110: 997-1005. [Crossref]
- Marina NM, Etcubanas E, Parham DM, Bowman LC, Green A (1989) Peripheral primitive neuroectodermal tumor (peripheral neuroepithelioma) in children. A review of the St Jude experience and controversies in diagnosis and management. Cancer 64: 1952-1960. [Crossref]
- Krithika CL, Sathasivasubramanian S, Vedeswari CP (2015) Peripheral Primitive Neuroectodermal Tumour of the Maxilla and Paranasal Sinuses: A Rare Clinical Presentation. J Clin Diagn Res 9: ZD32-ZD34. [Crossref]
- Mills SE (2002) Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinomas. Mod Pathol 15: 264-278. [Crossref]
- Pereira CM, de Abreu Alves F, Corrêa ME, Lima CS, de Almeida OP (2005) Mouth metastasis of peripheral primitive neuroectodermal tumor. Oral Dis 11: 44-45. [Crossref]
- Swanson PE, Humphrey PA, Dehner LP (1993) Immunoreactivity for bcl-2 protein in peripheral primitive neuroectodermal tumors. Appl Immunohistochem 1: 182-187.
- Jurgens H, Bier V (1988) Malignant peripheral neuroecto dermal tumors a retrospective analysis of 42 patients. Cancer 61: 349-357. [Crossref]
- Kushner BH, Meyers PA (2001) How effective is dose-intensive/ myeloablative therapy against Ewing’s sarcoma/primitive neuroectodermal tumour metastatic to bone or bone marrow? The memorial Sloan-Kettering experience and a literature review. J Clin Oncol 19: 870-880. [Crossref]
- Bhovi TV, Jaju PP, Singh A, Tiwari P (2015) Peripheral Primitive Neuroectodermal Tumor of the Maxilla. J Res Adv Dent 4: 1s: 203-207. [Crossref]
- Jayakumar S, Power D (2005) Ewing's Sarcoma/PNET: A Histopathological Review. IJOS 3.
- Al Haddabi I, Al Bahri M, Burney I (2012) Cytokeratin-positive primitive neuroectodermal tumor of the prostate: Case report and review of literature. Indian J Pathol Microbiol 55: 569-571. [Crossref]