Abstract
Metastasis is the leading cause of breast cancer-related mortality and involves cells from a primary tumor invading into adjacent tissue by using the circulatory system. During the process of metastasis, several dynamic and reciprocal interactions between cancer cells and inflammatory cells occur in the surrounding tumor microenvironment via the secretion of several cytokines, including tumor necrosis factor-α, and transforming growth factor-β. The present study aimed to investigate the functional role of recombinant apurinic apyrimidinic endonuclease 1(APE1)/Redox factor-1 (Ref-1) with reducing activity, [APE1/Ref-1-(SH)2] in the regulation of metastatic migration in cytokine-stimulated MDA-MB-231 breast cancer cells. The results reveal that [APE1/Ref-1-(SH)2], but not the oxidized form of recombinant APE1/Ref-1, attenuated migration, and invasion of cytokines-stimulated MDA-MB-231 cells. These effects were induced by the oxidative conformational changes due to the thiol-disulfide exchange reaction targeting disulfide bonds in extracellular domain of the cytokine receptors. Blocking of the binding of the cytokines to the corresponding receptors also reversed epithelial‐to-mesenchymal transition (EMT) in cytokine-stimulated MDA-MB-231 cells. The reversal of EMT was accompanied by changes in the expression levels of representative EMT markers, E-cadherin, N-cadherin, vimentin, and snail. Remarkable phenotypic changes were also observed in MDA-MB-231 cells, as the cells adopted an epithelial like globular shape, with regular dimension, a reorganized cytoskeleton, and collapsing filopodia and lamellipodia. In conclusion, although devising a stable form of recombinant [APE1/Ref-1-(SH)2] requires further study, its ability to target cytokine receptors and to inhibit their intracellular signaling suggests the use of [APE1/Ref-1-(SH)2] as a promising agent for regulating metastatic cancer cells.
Keywords
apurinic apyrimidinic endonuclease 1/Redox factor-1, reducing activity, cytokine receptor, metastasis, reverse epithelial‐to-mesenchymal transition
Introduction
Regulation of metastasis during tumor development has been discovered to represent an important therapeutic strategy [1]. Epithelial to mesenchymal transition (EMT) of tumor cells is the initial transformation step in the process of metastasis that is required to promote aggressive cell migration [2,3]. EMT is characterized by cellular phenotypic changes, more specifically, the acquisition of a mesenchymal phenotype, loss of epithelial polarity, and cell to cell contacts. All together these changes facilitates cell invasion and migratory abilities [3,4]. EMT is a reversible process, known as reverse EMT or mesenchymal to epithelial transition (MET), which was found to be induced by treatment with certain phytochemicals, such as resveratrol [5], pristimerin [6] or organic compounds [7-10]. A subpopulation of cells with reverse EMT properties have a retarded metastatic ability and an increased susceptibility to anticancer drug. Notably, in vivo studies have shown that some metastatic lesions and primary breast tumor counterparts share a similar epithelial properties under therapeutic conditions, which is less differentiated than the primary tumor [11,12].
Proinflammatory cytokines, such as and interleukin-1 [13], tumor necrosis factor-α (TNF-α) [14], and transforming growth factor-β (TGF-β) [15] in the tumor microenvironment can facilitate tumor progress and metastasis. In addition, they increase inflammatory signaling and innate immune responses by activating a plethora of downstream signaling pathways through the corresponding receptors. Therefore, regulating cytokine-mediated signaling has been investigated as a promising therapeutic target for malignant breast tumors. For example, there are numerous studies reporting the effect of monoclonal antibodies (infliximab, or adalimumab) in TNF-α blockade [16,17], etanercept fusion proteins [14,17], pan-TGF-β neutralizing antibody [15,18], P144 peptidic inhibitor [19], and the LY2109761 chemical inhibitor [15,20]. These studies demonstrated that inflammatory cytokines played a critical role in the aggressive metastatic behavior of breast cancer cells. Based on these aforementioned results, targeting the TNF-α/TGF-β inflammatory signaling pathway bears significant promise as a novel therapeutics for breast cancer treatment.
Our previous study reported that recombinant apurinic apyrimidinic endonuclease 1 (APE1)/Redox factor-1 (Ref-1) with reducing activity [APE1/Ref-1-(SH)2] promoted a conformational change in the TNF-α receptor 1 (TNFR1) inducing thiol-disulfide exchange, which regulated inflammatory signaling in TNF-α-stimulated vascular endothelial cells [21]. The APE1/Ref-1 protein has demonstrated both redox and DNA repair activities in all cell types [22]. Notably, previous studies, including ours, observed that APE1/Ref-1 was released extracellularly in hyperacetylated cells [21,23,24] as well as systemic lupus erythematosus [25], lung cancer [26], bladder cancer [27], and under conditions of endotoxemia [28]. Furthermore, the secreted APE1/Ref-1 from hyperacetylated breast cancer cells modulated metastasis, tumor growth and proliferation in vitro [24] and in vivo [29].
The present study was performed to determine the functional role of reduced form of recombinant APE1/Ref-1, as a new inhibitory candidate of TNF-α/TGF-β signaling. [APE1/Ref-1-(SH)2] exhibited significant inhibitory effects over migration in MDA-MB-231 cells stimulated with TNF-α/TGF-β. The expression levels of EMT-related proteins were also altered, alongside the occurrence of reverse EMT-induced morphological change. These results suggested that [APE1/Ref-1-(SH)2] may regulate the migration of breast cancer cells by promoting thiol-disulfide exchange reaction in the TNFR and TGF-β receptor (TGFR), which may underlie the anti-metastatic activity of [APE1/Ref-1-(SH)2].
Materials and methods
Cells, antibodies, and reagents. Triple negative mesenchymal-like human breast cancer cell lines MDA-MB-231 (HTB-26) and BT-549 (HTB-122) [30] were obtained from American Type Culture Collection. The cell lines were confirmed by short tandem-repeat profiling and assessment of cellular morphology. RPMI-1640 medium (11875093), penicillin-streptomycin antibiotics (15140122) and fetal bovine serum (FBS, 16000044) were obtained from Gibco, Thermo Fisher Scientific. Human TNF-α (H8916), TGF-β (T1654), Trichostatin A (TSA, T1952), and acetyl salicylic acid (ASA, S5922) were purchased from Sigma Aldrich. Antibodies against E-cadherin (4A2, 14472), N-cadherin (13A9, 1425), vimentin (5G3F10, 3390), snail (L70G2, 3895), and β-actin (8H10D10, 3700) were purchased from Cell Signaling Technology. The Cultrex basement membrane extract (BME, 3432-001-01) cell invasion assay kit was from Trevigen.
Cell culture. The MDA-MB-231 human breast adenocarcinoma cells, a mesenchymal breast cancer model, were cultured at 37°C with 5% CO2 in RPMI 1640 medium with 10% FBS and 1% antibiotics. For providing hyperacetylation condition, the MDA-MB 231 cells were treated with 0.5 μM TSA for 1 h and then treated with different concentrations of ASA. Some cells were pretreated with [APE1/Ref-1-(SH)2] or oxidized form of recombinant APE1/Ref-1, [APE1/Ref-1-S2] for 1h and then stimulated with cytokines, including 20 ng/ml TNF-α, and/or 10 ng/ml TGF-β prior to further incubation for the indicated duration. The morphological changes in the treated cells were observed under an inverted phase-contrast light microscope (Leica Microsystems GmbH, Germany).
EMT scores was calculated by counting the number of the mesenchymal-like and epithelial-like cells [31]. Five randomly selected fields were chosen and the cells in each field were counted at an average about 70% confluence. It was replicated by two independent experimenters.
Preparation of [APE1/Ref-1-(SH)2]. The pure form of [APE1/Ref-1-(SH)2] was prepared as previously reported [28]. To test the reducing activity of recombinant APE1/Ref-1, a biochemical method using NAD(P)H-Glo Detection System (Promega, Corporation, WI, USA) was adopted with some modification [21]. The recombinant [APE1/Ref-1-(SH)2] with the addition of 1 mM dithiothreitol (DTT) waws maintained in liquid nitrogen tank. When [APE1/Ref-1-(SH)2] was used, DTT was removed using the spin column, then [APE1/Ref-1-(SH)2] was diluted in neutralization buffer (20 mM HEPES, 100 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, pH 7.7).
Cell migration assay by wound-healing. Cells were plated into a 6-well plate and incubated until they formed more than 90 % confluent monolayer. Cells underwent serum-starvation for 24 h and the monolayer was subsequently scratched with a pipette tip; the nonadherent cells were removed by washing the plate with serum-free RPMI1640 medium. Cells were subsequently treated with TNF-α (20 ng/mL) or TGF-β (10 ng/mL) along with either 1 μg/mL [APE1/Ref-1-(SH)2] or [APE1/Ref1-S2] for 24 h. During this time, the cells were permitted to migrate, and where then visualized in three randomly selected fields of view per well through an inverted light microscope (magnification, x100). The number of cells migrating into the wound area was analyzed using Image J software (National Institute of Health, http://rsb.info.nih.gov/ij/).
Invasion assay. Cultrex ® 96-well basement membrane extract (BME) cell invasion assay kit was used for our invasion assay. Transwell chambers with 8-μm pore polycarbonate filters were treated with 0.8x BME in coat buffer and incubated for 24 h at 37°C. Following coating of the chambers, cells (1×105) were resuspended in RPMI 1640-medium with various concentrations of [APE1/Ref-1-(SH)2] and plated onto the BME-coated filter in the upper chamber of the insert, which contained serum-free medium. The lower chamber was filled with medium containing TNF-α (20 ng/mL) or TGF-β (10 ng/mL); the control wells only contained media. Following incubation for 24 h, the migratory cells on the bottom of the membrane were stained with Calcein-AM and counted using a fluorometer (Promega Corporation, WI, USA).
Immunoblotting. Cells were harvested and lysed following the previous protocol [24]. The cell lysate was centrifuged at 12,000 x g for 20 min at 4℃, and the supernatant was used for immunoblotting. After protein quantification, equal amounts of protein were separated via SDS-PAGE and subsequently transferred onto PVDF membrane. The membrane was then blocked and incubated with specific primary antibodies for E-cadherin, N-cadherin, vimentin, snail, and anti-β-actin. The membranes were then incubated with a secondary antibody and the protein bands were visualized, and the band intensity was quantified using a densitometer and normalized by β-actin control.
Statistical analysis. Statistical differences between the control and [APE1/Ref-1-(SH)2]-treated groups were determined using one-way analysis of variance (ANOVA). Dunnett’ or Tukey’s correction post hoc test was used for multiple comparisons using GraphPad (Prism 9) software considering P<0.05 as statistical significance.
Results
Hyperacetylation induces mesechymal-epithelial like morphological changes in MDA-MB‑231 cells.
Our previou studies reported that hyperacetylation, induced by treatment with 0.5 μM TSA and various concentrations of ASA inhibited on viability and proliferation of triple negative breast cancer (TNBC) cells including MDA-MB-231 cells [24,29]. Therefore, it was plausible that the apoptotic cell death of the aggressive TNBC cell line, MDA-MB-231, following hyperacetylation was linked to an increased susceptibility to anti-cancer drugs, and the reversal of EMT [32-34]. To verify the effects of hyperacetylation on reverse of EMT, MDA-MB‑231 cells were pre-exposed to 0.5 μM TSA and then further treated with 1mM ASA for 24 h. The morphology of MDA-MB‑231 cells was altered in response to TSA treatment (Figure 1A, middle panel). The changes in hyperacetylation were observed under phase contrast microscopy, and the cells were observed to be shifted from an elongated spindle-like shape, with pronounced cellular scattering due to the presence of filopodia and lamellipodia, to a more globular epithelial phenotype, which formed sheet-like monolayers, and permitted the formation of cell-cell junctions (Figure 1A, left) [12,35,36]. EMT score (%) were calculated by counting mesenchymal or epithelial-like cells and dividing them with total cell numbers [31]. The number of epithelial cells was increased approximately ~1.84 folds in hyperacetylation condition compared with the control cells (Figure 1A, right bar graph).
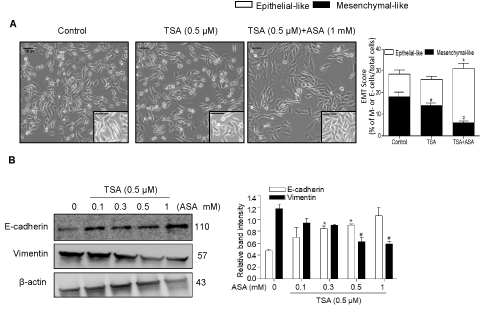
Figure 1: Hyperacetylation induces morphological changes in MDA-MB-231 cells and regulates mesenchymal-epithelial transition associated molecules. (A) MDA-MB-231 cells were pretreated with 0.5 μM trichostatin A for 1 h and then acetylsalicylic acid at the indicated concentration for 24 h. Representative morphology of the cells under an inverted microscope are shown (magnification x100; inset, 1.5-fold magnification). Scale bar represents 100 μm. Bar graph shows mean EMT score by counting of mesenchymal-like or epithelial-like cells independent scoring results. Data are presented as % mesenchymal like- or epithelial like cells. Columns, mean (n = 10); bars, ± SEM. Data were analyzed using a one-way ANOVA followed by Dunnett’s post hoc test. *, # P<0.05 vs. control cells. (B) The expression levels of E-cadherin and vimentin were analyzed using immunoblotting assay. The blots were stripped and re-probed with anti-actin antibody to confirm protein loading. Representative blots are shown. The bar graph shows the densitometric semi-quantification of the immunoblottings. Data are presented as % densitometry values of E-cadherin and vimentin expression levels. Columns, mean (n = 3); bars, ± SEM. Data were analyzed using a one-way ANOVA followed by Dunnett’s post hoc test. *, # P<0.05 vs. control cells.
As hyperacetylation resulted in marked mophological changes in MDA-MB-231 cells, the expression levels of the EMT representative markers such as E-cadherin and vimentin were analyzed. As shown in (Figure 1B), immunoblot analysis revealed that the expression levels of E-cadherin were increased following hyperacetylation. The expression levels of vimentin were inversely associated with the expression levels of E-cadherin, indicating that EMT was reversed. In more detail, the expression levels of E-cadherin were shown to be upregulated following a 24 h exposure to 0.5 μM TSA and ASA in a concentration-dependent manner. In contrast, the expression levels of vimentin were downregulated by ~53.2% following hyperacetylation compared with the control cells. These results suggest that hyperacetylation may reverse the EMT process in MDA-MB‑231 cells, which would subsequently induce the loss of metastatic potential.
Treatment of recombinant [APE1/Ref-1-(SH)2] reverses the EMT-like morphological and molecular changes in cytokine-stimulated MDA-MB-231 cells.
Next, it was determined whether treatment with [APE1/Ref-1-(SH)2] could reverse EMT via its redox function and thereby regulate TNF-α or TGF-β-mediated metastatic signalings. The morphological changes of MDA-MB-231 cells following stimulation with TNF-α or TGF-β cytokines were first observed. The mesenchymal morphology of MDA-MB-231 cells was more pronounced following exposure to TNF-α or TGF-β cytokines, with cells exhibiting a spindle like, flattened, elongated shape with lamellipodia and filopodia (Figure 2A, left). However, the pretreatment of cells with [APE1/Ref-1-(SH)2] induced significant morphological changes in MDA-MB-231 cells, even following stimulation with TNF-α or TGF-β cytokines, with cells exhibiting an epithelial-like polygonal shape with regular dimensions, a reorganized cytoskeletons and collapsing poidas. These mophological changes occured in a concentration-dependent manner (Figure 2A). The epithelial subpopulation in pretreated cells with [APE1/Ref-1-(SH)2] was increased ~3.65 and ~3.19 folds with TNF-α and TGF-β respectively, compared with the ones treated with either cytokine alone.
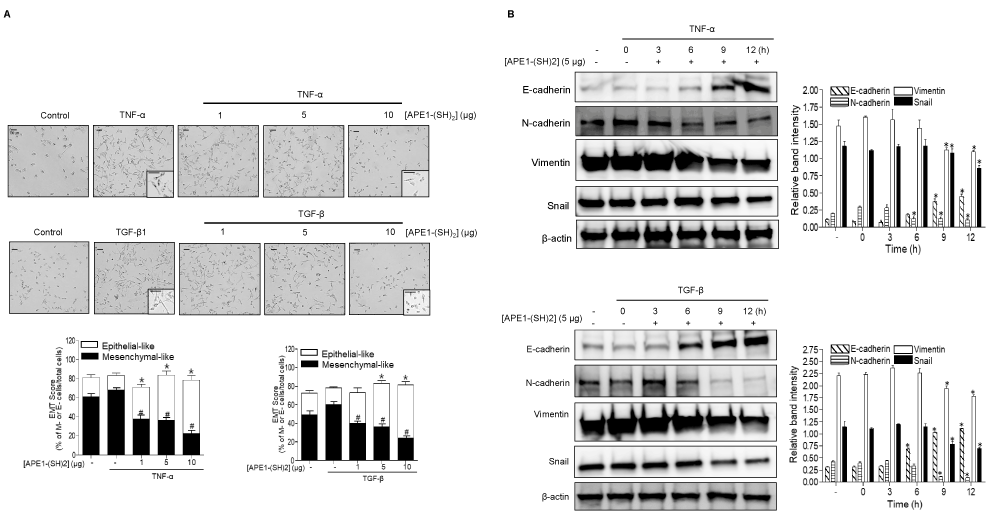
Figure 2: Pretreatment with [APE1/Ref-1-(SH)2] causes reverse EMT of MDA-MB-231 cells even following TNFα-/TGF-β-stimulation. MDA-MB-231 cells were pretreated [APE1/Ref-1-(SH)2] at the indicated concentration for 2 h, followed by further incubation with either 20 ng/ml TNF-α (upper) or 10 ng/ml TGF-β (lower) for 18 h. (A) Morphological changes associated with reverse EMT were shown using phase‐contrast microscopy (magnification, x100; inset, 2-fold magnification). Scale bar represents 100 μm. The cells were elongated and spindle‐shaped with an increased intercellular separation and pseudopodia, following the treatment with TNF-α or TGF-β, but not in cells pretreated with [APE1/Ref-1-(SH)2]. The bar graph shows mean EMT score. Columns, mean (n = 6); bars, ± SEM. Data analyzed using a one-way ANOVA followed by Dunnett’s post hoc test. *, # P<0.05 vs. cytokine-stimulated control cells. (B) Immunoblot analysis of expression levels of EMT representative markers was performed using MDA-MB-231 cell lysate. The blots were stripped and re-probed with an anti-β-actin antibody to confirm equal protein loading. Representative blots are shown. The bar graph shows the densitometric semi-quantification of the immunoblotting data in the order of E-cadherin, N-cadherin, Vimentin and Snail. Columns, mean (n = 3); bars, ±SEM. Data analyzed using a one-way ANOVA followed by a Dunnett’s post hoc test. *P<0.05 vs. TNF-α, or TGF-β-treated control cells.
Consistent with the morphological changes of TNF-α- or TGF-β-stimulated MDA-MB-231 cells, the expression levels of E-cadherin, N-cadherin, vimentin and snail were changed in a time-dependent manner, and the results were similar to the observed effects of hyperacetylated cells on reverse EMT (Figure 2B). The expression of E-cadherin was upregulated by ~ 7.67- or 2.67-fold following pre-exposure to [APE1/Ref-1-(SH)2] and then TNF-α or TGF-β-stimulation, respectively compared with cytokine-treated control cells. Conversely, the expression levels of other EMT markers such as N-cadherin, vimentin and snail were significantly downregulated following pre-exposure to [APE1/Ref-1-(SH)2] and TNF-α or TGF-β-stimulation. Similar results were obtained with BT-549 cells, another triple negative breatst cancer cell line (Figure S1). Both the observed morphological and molecular changes suggested that MDA-MB-231 cells may undergo reverse EMT following the pre-treatment with [APE1/Ref-1-(SH)2], even in the presence of inflammatory cytokines.
Pretreatment of [APE1/Ref-1-(SH)2] inhibits the migration of cytokine-stimulated MDA-MB‑231 cells. To further determine the functional role of the [APE1/Ref-1-(SH)2]-induced reversal of EMT, the redox effect on the migration of MDA-MB-231 cells stimulated with TNF-α and TGF-β was investigated. The cell monolayers were pretreated with [APE1/Ref-1-(SH)2] and then scratched for the wound healing assay. The pretreatment with [APE1/Ref-1-S2] promoted the migration of cells (Figure 3A, top panel). However, [APE1/Ref-1-(SH)2] treatment significantly inhibited MDA-MB‑231 cells from migration, even following TNF-α and TGF-β stimulation (Figure 3A, bottom panel). For example, the wound area of cells pretreatd with [APE1/Ref-1-(SH)2] was reduced to ~17.4 %, while the migration of cytokine-stimulated cell was recoverd by ~ 43.6 % following the treatment with [APE1/Ref-1-S2]. The recovered wound area of [APE1/Ref-1-S2]- treated cells was significantly increased in a time-dependent manner, but not in cells treated with [APE1/Ref-1-(SH)2] (Figure 3B). These data suggested that [APE1/Ref-1-(SH)2] may abrogate TNF-α/TNFR, or TGF-β/TGFR signaling, thereby inhibiting the migration of cytokine-stimulated MDA-MB-231 cells.
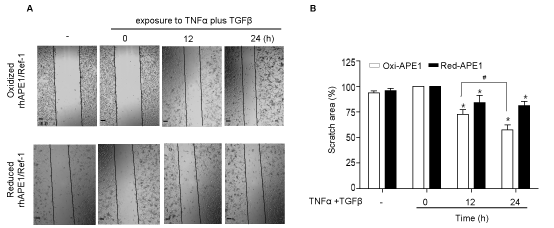
Figure 3: [APE1/Ref-1-(SH)2] inhibits the migration of MDA-MB-231 cells stimulated with cytokine. (A) Confluent monolayers of MDA-MB-231cells were scratched and then pretreated with recombinant [APE1/Ref-1-S2] or [APE1/Ref-1-(SH)2], followed by 20 ng/ml TNF-α and 10 ng/ml TGF-β. A phase contrast microscope (magnification, x100) was used to observe cell migration into the wound area at the indicated durations. Scale bar represents 100 μm. Representative images of the wound-healing assay at different durations are shown. (B) Semi-quantification of the number of migratory cells was obtained from three separate experiments. The wound area was analyzed using Image J software. Data are expressed as the % of a time-dependent differences following exposure to TNF-α and TGF-β. Columns, mean (n = 3); bars, ±SEM. Data analyzed using a one-way ANOVA followed by a Dunnett’s post hoc test. * P<0.05 vs. TNF-α plus TGF-β-treated cells; # P<0.05 vs. recombinant protein-treated cells.
[APE1/Ref-1-(SH)2] attenuates the invasion of cytokine-stimulated MDA-MB 231 cells. As cell invasion is also indispensable for tumor metastasis [1,2,4], the effect of [APE1/Ref-1-(SH)2] on cell invasion was evaluated using a BME transwell assay. The transwell invasion system comprised an upper chamber with a porous membrane and a lower well that contained TNF-α, or TGF-β. MDA-MB-231 cells were plated into the upper chamber with [APE1/Ref-1-(SH)2] and allowed to invade through the pores to the other side of the membrane (Figure 4A). The invasive activity of MDA-MB‑231 cells was significantly attenuatted following [APE1/Ref-1-(SH)2] treatment in a dose-dependent manner (Figure 4B); the number of invasive MDA-MB-231 cells following [APE1/Ref-1-(SH)2] treatment was decreased by ~42% compared with the control cells treated with only TNF-α (Figure 4B, left panel). Cell invasion was not statistically significantly inhibited in cells treated with [APE1/Ref-1-(SH)2] and TGF-β compared with the cells only stimulated TGF-β (Figure 4B, right panel). These results suggest that the redox activity of [APE1/Ref-1-(SH)2] may have an important role in thiol-disulfide exchange reactions for TNFR1 or TGFR, which promotes the conformational change of their extracellular domain, and leads to the regulation of TNF-α or TGF-β-stimulated signaling [21,37].
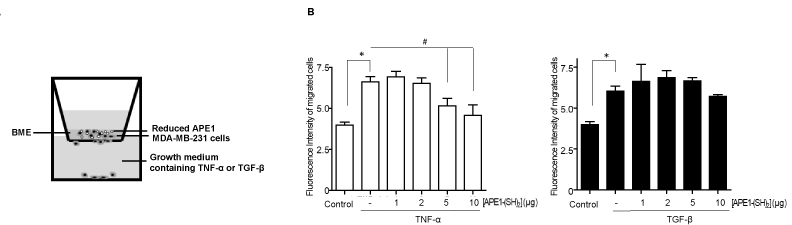
Figure 4: [APE1/Ref-1-(SH)2] attenuates the invasion of MDA-MB-231 cells stimulated with cytokine. (A) Schematic representation of the transwell invasion assay. MDA-MB‑231 cells were pretreated with various concentrations of [APE1/Ref-1-(SH)2] and plated on a base membrane extract-coated membrane (8-μm), and the upper chamber contained either 20 ng/ml TNF-α, or 10 ng/ml TGF-β. (B) Fluorescent intensity of the cells invading the membrane was measured. Columns, mean (n = 3); bars, ±SEM. Data analyzed using a one-way ANOVA followed by a Dunnett’s post hoc test. * P<0.05 vs. control cells; # P<0.05 vs. TNF-α-treated cells.
Discussion
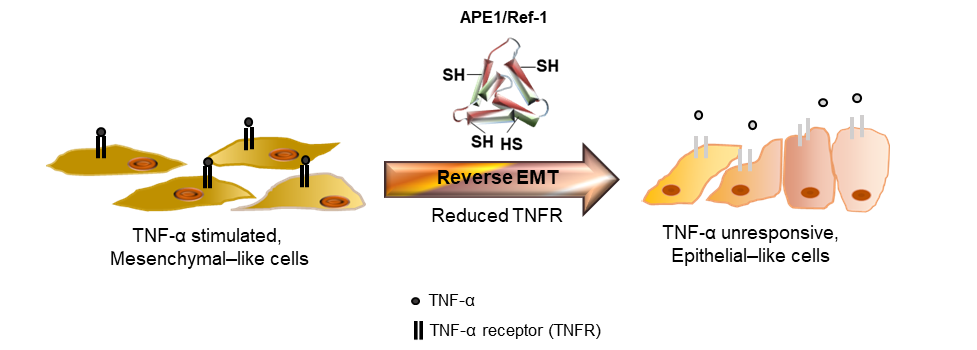
Due to the damage to normal cells or drug resistance in following cancer treatment, targeted therapeutic proteins, including peptides, have certain advantages, such as specificity, higher affinity, safety and less drug-drug interactions compared with chemical compounds [38]. Another advantage of using proteins as therapeutic drugs is the minimal toxic side effects, as they do not accumulate in kidney or liver [38]. For example, an apoptosis-inducing ligand related to TNF is an integral membrane protein that binds to its receptors on the cell surface of cancer cell and leads to apoptotic cell death and it has been tested in clinics as a potential therapeutic protein [39]. In addition, genetically modifed recombinant proteins corresponding to IFN-α and chimeric immunotoxins, and tumor-specific ligands linked to a modified toxin have also been widely proposed as potential adjuvant therapies for the treatment of various tumors types in vitro and in vivo [40-42]. Consistent with the previous studies, the present study demonstrated the potential of extracellularly secreted APE/Ref-1 as a therapeutic protein for treating TNBC cells [21,24,29]. Furthermore, in the current study, recombinant [APE1/Ref-1-(SH)2] regulated the migration and invasion of MDA-MB-231 cells stimulated with TNF-α or TGF-β. The underlying mechanism of [APE1/Ref-1-(SH)2] was suggested to involve its reducing activity of the disulfide bonds in the extracellular domain of TNFR, which has 12 disulfide bonds, to block its subsequent signalings (Figure 5). Therefore, the APE1/Ref-1 protein may represent a novel and promising therapeutic candidate for the regulation of tumor metastasis and apoptotic cell death via targeting TNFR or TGFR on the immune cells surrounding the tumor.
Figure 5: Schematic diagram of potential of [APE1/Ref-1-(SH)2] which may cause reverse EMT in TNF-α-stimulated MDA-MB-231 cells. Based on the present study using [APE1/Ref-1-(SH)2] with reducing activity and anti-inflammatory function [21], we propose that [APE1/Ref-1-(SH)2] causes oxidative conformational changes in TNF receptors (TNFR). Their thiol-disulfide exchange reaction may target disulfide bonds in extracellular domain of the TNFR on TNF-α-stimulated, mesenchymal-like MDA-MB-231 cells. The [APE1/Ref-1-(SH)2]-induced reversed EMT with retarded metastatic migration was accompanied with the changes of expression of EMT markers, and phenotypic changes even in the presence of TNF-α.
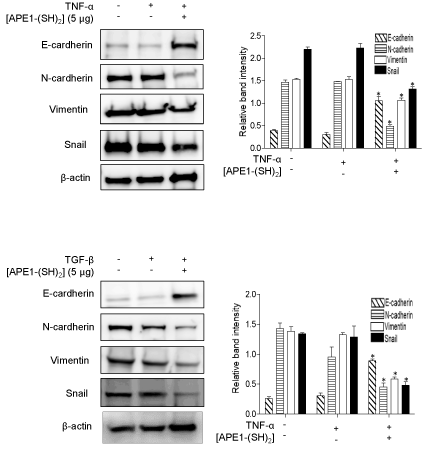
Figure S1: Immunoblot analysis of expression levels of EMT representative markers was performed using the cell lysate of BT-549. Representative blots are shown. The bar graph shows the densitometric semi-quantification of the immunoblotting data. Columns, mean (n = 3); bars, ±SEM. Data analyzed using a one-way ANOVA followed by a Dunnett’s post hoc test. *P<0.05 vs. TNF-α or TGF-β-treated control cells.
However, the application of therapeutic proteins remains a significant challenge when considering how to maintain a stable structure alongside effective activity. In the presnet study, [APE1/Ref-1-(SH)2] was purified and then maintained in DTT to conserve the free sulfhydryl groups as a commercially available protein. Immediately before use, the excess DTT was removed so that the [APE1/Ref-1-(SH)2] could exert its full reducing activity. However, the short half-life of [APE1/Ref-1-(SH)2] resulted from oxidation and the unknown effects of [APE1/Ref-1-(SH)2] in the presence of remnant preservatives, are still issues of [APE1/Ref-1-(SH)2] use and need to be addressed [21]. Therapeutic peptide, which is usually <50 amino acids in length, exerted its activity via (SH)2 in the catalytic domain. For examples, the human neutrophil peptide-1 has several cysteine residues and exerted an anti-cancer effect on oral squamous cells [43] and PC-3 prostate cancer cells [44]. Another approach to maintain the activity and half-life of therapeutic proteins involves the conjugation of polyethylene glycol (PEG) to proteins, although some challenges, such as the induced increase in the macro-size/length of the protein and clearance of protein through renal filtration in vivo have emerged [38,45]. We have established conditions which pegylated-[APE1/Ref-1-(SH)2] exhibits reducing activity. The anticancer effects of PEG-[APE1/Ref-1-(SH)2] have been further analyzed in various cancer cells types.
Cancer cells undergo a cellular process called EMT. During this event, epithelial cells acquire mesenchymal phenotypes and metastatic behaviors [3,4]. In cancer cells, regressive changes in E-cadherin expression alongside the acquiration of a mesenchymal morphology are associated with an increased metastatic potential and tumor grade [46]. The strong migratory and invasive abilities of cancer cells lead to the metastasis of cancer cells from the original site to distant organs through blood and lymphatic vessels [2,3]. In addition, upregulated vimentin expression and increased intermediate filaments maintain the integrity of mesenchymal cells. The expression levels of vimentin were found to be significantly upregulated in high-grade ductal breast carcinoma [2]. Inverse changes of up-regulated E-cadherin and down-regulated vimentin in the expression levels are representative molecular markers of reverse EMT. Therefore, the ability of [APE1/Ref-1-(SH)2] to reverse EMT in MDA-MB-231 cells was hypothesized to occur via the regulation of E-cadherin, N-cadherin, vimentin and snail expression levels, which inhibited the migration and invasion of MDA-MB-231 cells, even following cytokine-stimulation. However, there were still some cells in the reverse EMT condition that migrated to the lower chamber of our invasion assay indicating that [APE1/Ref-1-(SH)2] didn’t affect the subset of cells leaving them maintaining characteristics of mesenchymal cells. This result reminds us of the above mentioned challenge in using [APE1/Ref-1-(SH)2] in future therapeutics. It is possible that [APE1/Ref-1-(SH)2] was not stable enough in the assay by itself to synchronously induce reverse EMT for prolonged time.
Conclusion
The findings of the present study suggested that [APE1/Ref-1-(SH)2] may inhibit the migration and invasion of cytokine-stimulated tumor cells. These may occur by inhibiting TNFR and TGFR signalings and revesing EMT. These results indicated that recombinant [APE1/Ref-1-(SH)2] may have potential to be further developed as an effective drug candidate to target the invasion and migration of aggressive tumor cells, like TNBC cells, by re-programming the tumor microenvironment.
Acknowledgments
We thank Prof. B.H. Jeon of the Chungnam National University for the generous gift of an APE1/Ref-1 construct.
Funding
This study was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A1B01009752 S.C).
Conflicts of interest
The authors declare that they have no competing interests.
Reference
- 1. Steeg PS, Theodorescu D (2008) Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol 5: 206-219. [Crossref]
- 2. Thompson EW, Newgreen DF, Tarin D (2005) Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res 65: 5991-5995. [Crossref]
- 3. Yao D, Dai C, Peng S (2011) Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol Cancer Res 9: 1608-1620. [Crossref]
- 4. Skovierova H, Okajcekova T, Strnadel J, Vidomanova E, Halasova E (2018) Molecular regulation of epithelial-to-mesenchymal transition in tumorigenesis (Review). Int J Mol Med 41: 1187-1200. [Crossref]
- 5. Sun Y, Zhou QM, Lu YY, Zhang H, Chen QL, et al. (2019) Resveratrol Inhibits the Migration and Metastasis of MDA-MB-231 Human Breast Cancer by Reversing TGF-beta1-Induced Epithelial-Mesenchymal Transition. Molecules 24: 1131. [Crossref]
- 6. Li J, Guo Q, Lei X, Zhang L, Su C, et al. (2020) Pristimerin induces apoptosis and inhibits proliferation, migration in H1299 Lung Cancer Cells. J Cancer 11: 6348-6355. [Crossref]
- 7. Lu A, Wang W, Wang-Renault SF, Ring BZ, Tanaka Y, et al. (2020) 5-Aza-2'-deoxycytidine advances the epithelial-mesenchymal transition of breast cancer cells by demethylating Sipa1 promoter-proximal elements. J Cell Sci 133: jcs236125. [Crossref]
- 8. Mali AV, Joshi AA, Hegde MV, Kadam SS (2018) Enterolactone modulates the ERK/NF-kappaB/Snail signaling pathway in triple-negative breast cancer cell line MDA-MB-231 to revert the TGF-beta-induced epithelial-mesenchymal transition. Cancer Biol Med 15: 137-156. [Crossref]
- 9. Vultur A, Buettner R, Kowolik C, Liang W, Smith D, et al. (2008) SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol Cancer Ther 7: 1185-1194. [Crossref]
- 10. Yu JK, Yue CH, Pan YR, Chiu YW, Liu JY, et al. (2018) Isochlorogenic Acid C Reverses Epithelial-Mesenchymal Transition via Down-regulation of EGFR Pathway in MDA-MB-231 cells. Anticancer Res 38: 2127-2135. [Crossref]
- 11. Kowalski PJ, Rubin MA, Kleer CG (2003) E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res 5: R217-222. [Crossref]
- 12. Chao YL, Shepard CR, Wells A (2010) Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer 9: 179. [Crossref]
- 13. Esquivel-Velazquez M, Ostoa-Saloma P, Palacios-Arreola MI, Nava-Castro KE, Castro JI, et al. (2015) The role of cytokines in breast cancer development and progression. J Interferon Cytokine Res 35: 1-16. [Crossref]
- 14. Madhusudan S, Foster M, Muthuramalingam SR, Braybrooke JP, Wilner S, et al. (2004) A phase II study of etanercept (Enbrel), a tumor necrosis factor alpha inhibitor in patients with metastatic breast cancer. Clin Cancer Res 10: 6528-6534. [Crossref]
- 15. Ganapathy V, Ge R, Grazioli A, Xie W, Banach-Petrosky W, et al. (2010) Targeting the Transforming Growth Factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol Cancer 9: 122. [Crossref]
- 16. Hamaguchi T, Wakabayashi H, Matsumine A, Sudo A, Uchida A (2011) TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem Biophys Res Commun 407: 525-530. [Crossref]
- 17. Mercogliano MF, Bruni S, Elizalde PV, Schillaci R (2020) Tumor Necrosis Factor alpha Blockade: An Opportunity to Tackle Breast Cancer. Front Oncol 10: 584. [Crossref]
- 18. Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, et al. (2012) TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U S A 109: 16618-16623. [Crossref]
- 19. Llopiz D, Dotor J, Casares N, Bezunartea J, Diaz-Valdes N, et al. (2009) Peptide inhibitors of transforming growth factor-beta enhance the efficacy of antitumor immunotherapy. Int J Cancer 125: 2614-2623. [Crossref]
- 20. Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, et al. (2008) LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther 7: 829-840. [Crossref]
- 21. Park MS, Choi S, Lee YR, Joo HK, Kang G, et al. (2016) Secreted APE1/Ref-1 inhibits TNF-alpha-stimulated endothelial inflammation via thiol-disulfide exchange in TNF receptor. Sci Rep 6: 23015. [Crossref]
- 22. Pines A, Perrone L, Bivi N, Romanello M, Damante G, et al. (2005) Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res 33: 4379-4394. [Crossref]
- 23. Choi S, Lee YR, Park MS, Joo HK, Cho EJ, et al. (2013) Histone deacetylases inhibitor trichostatin A modulates the extracellular release of APE1/Ref-1. Biochem Biophys Res Commun 435: 403-407. [Crossref]
- 24. Lee YR, Kim KM, Jeon BH, Choi S (2015) Extracellularly secreted APE1/Ref-1 triggers apoptosis in triple-negative breast cancer cells via RAGE binding, which is mediated through acetylation. Oncotarget 6: 23383-23398. [Crossref]
- 25. Katsumata Y, Kawaguchi Y, Baba S, Hattori S, Tahara K, et al. (2011) Identification of three new autoantibodies associated with systemic lupus erythematosus using two proteomic approaches. Mol Cell Proteomics 10: M110 005330. [Crossref]
- 26. Dai N, Cao XJ, Li MX, Qing Y, Liao L, et al. (2013) Serum APE1 autoantibodies: a novel potential tumor marker and predictor of chemotherapeutic efficacy in non-small cell lung cancer. PLoS One 8: e58001. [Crossref]
- 27. Shin JH, Choi S, Lee YR, Park MS, Na YG, et al. (2015) APE1/Ref-1 as a Serological Biomarker for the Detection of Bladder Cancer. Cancer Res Treat 47: 823-833. [Crossref]
- 28. Park MS, Lee YR, Choi S, Joo HK, Cho EJ, et al. (2013) Identification of plasma APE1/Ref-1 in lipopolysaccharide-induced endotoxemic rats: implication of serological biomarker for an endotoxemia. Biochem Biophys Res Commun 435: 621-626. [Crossref]
- 29. Lee YR, Park MS, Joo HK, Kim KM, Kim J, et al. (2018) Therapeutic positioning of secretory acetylated APE1/Ref-1 requirement for suppression of tumor growth in triple-negative breast cancer in vivo. Sci Rep 8: 8701. [Crossref]
- 30. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, et al. (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121: 2750-2767. [Crossref]
- 31. Liu X, Li J, Cadilha BL, Markota A, Voigt C, et al. (2019) Epithelial-type systemic breast carcinoma cells with a restricted mesenchymal transition are a major source of metastasis. Sci Adv 5: eaav4275. [Crossref]
- 32. Lee AF, Chen MC, Chen CJ, Yang CJ, Huang MS, et al. (2017) Reverse epithelial-mesenchymal transition contributes to the regain of drug sensitivity in tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. PLoS One 12: e0180383. [Crossref]
- 33. Akbar MW, Isbilen M, Belder N, Canli SD, Kucukkaraduman B, et al. (2020) A Stemness and EMT Based Gene Expression Signature Identifies Phenotypic Plasticity and is A Predictive but Not Prognostic Biomarker for Breast Cancer. J Cancer 11: 949-961. [Crossref]
- 34. Xu J, Liu D, Niu H, Zhu G, Xu Y, et al. (2017) Resveratrol reverses Doxorubicin resistance by inhibiting epithelial-mesenchymal transition (EMT) through modulating PTEN/Akt signaling pathway in gastric cancer. J Exp Clin Cancer Res 36: 19. [Crossref]
- 35. Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, et al. (2011) FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res 71: 3076-3086. [Crossref]
- 36. Liu X, Feng R (2010) Inhibition of epithelial to mesenchymal transition in metastatic breast carcinoma cells by c-Src suppression. Acta Biochim Biophys Sin (Shanghai) 42: 496-501. [Crossref]
- 37. Joo HK, Lee YR, Lee EO, Park MS, Choi S, et al. (2019) The extracellular role of Ref-1 as anti-inflammatory function in lipopolysaccharide-induced septic mice. Free Radic Biol Med 139: 16-23. [Crossref]
- 38. Marqus S, Pirogova E, Piva TJ (2017) Evaluation of the use of therapeutic peptides for cancer treatment. J Biomed Sci 24: 21. [Crossref]
- 39. Rahman M, Pumphrey JG, Lipkowitz S (2009) The TRAIL to targeted therapy of breast cancer. Adv Cancer Res 103: 43-73. [Crossref]
- 40. Sabel MS, Sondak VK (2003) Pros and cons of adjuvant interferon in the treatment of melanoma. Oncologist 8: 451-458. [Crossref]
- 41. Li M, Rao C, Pei D, Wang L, Li Y, et al. (2014) Novaferon, a novel recombinant protein produced by DNA-shuffling of IFN-alpha, shows antitumor effect in vitro and in vivo. Cancer Cell Int 14: 8. [Crossref]
- 42. Choudhary S, Mathew M, Verma RS (2011) Therapeutic potential of anticancer immunotoxins. Drug Discov Today 16: 495-503. [Crossref]
- 43. McKeown ST, Lundy FT, Nelson J, Lockhart D, Irwin CR, et al. (2006) The cytotoxic effects of human neutrophil peptide-1 (HNP1) and lactoferrin on oral squamous cell carcinoma (OSCC) in vitro. Oral Oncol 42: 685-690. [Crossref]
- 44. Gaspar D, Freire JM, Pacheco TR, Barata JT, Castanho MA (2015) Apoptotic human neutrophil peptide-1 anti-tumor activity revealed by cellular biomechanics. Biochim Biophys Acta 1853: 308-316. [Crossref]
- 45. Roberts MJ, Bentley MD, Harris JM (2002) Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev 54: 459-476. [Crossref]
- 46. Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, et al. (2002) Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene 21: 3241-3246. [Crossref]