Thalidomide, first developed in Germany in 1956, is infamously known for its teratogenic properties when used for hyperemesis gravidarum and sedation [1]. From the late 1950s until 1962, use of this drug rapidly expanded to 48 countries where it was responsible for birth defects ranging from congenital heart disease, malformations in limb development and ocular abnormalities in thousands of children. While initial preclinical data suggested that thalidomide had an acceptable safety profile in rodents, testing in pregnant animals was not a mandated practice at the time [2]. The tragedy of thalidomide-rapidly led to significant changes in the processes required for medical product development throughout the world with enactment of an amendment to the United States Federal Food, Drug and Cosmetic Act in 1962 requiring more rigorous preclinical testing for new pharmaceuticals [3].
In the early 2000s, thalidomide was repurposed in the clinic as a potent antiangiogenic [4] and immunomodulatory compound [5] for the treatment of multiple myeloma (MM), myelodysplastic syndrome harboring a chromosomal deletion in 5q [del (5q) MDS] and other hematological malignancies [6,7]. The drug was approved for cancer treatment under an FDA directive requiring routine pregnancy testing with distribution of written and verbal warnings to inform patients of the detrimental impact of thalidomide on fetal development [8]. Many studies have confirmed the relationship between thalidomide, and its related class of compounds (also known as immunomodulatory drugs, IMiD®) and teratogenicity in humans, zebrafish, chickens and in rodents when given during specific periods of gestation [1,9-12]
Fifty years after the thalidomide tragedy, Takumi Ito and colleagues revealed the molecular mechanism of this drug through elegant studies conducted in zebrafish and chickens [12]. Cereblon (CRBN) was identified as the direct target of immunomodulatory compounds and one of many DDB1 and CUL4-associated factors (DCAFs) that directs protein turnover by ubiquitination and subsequent proteasome recognition. Cereblon acts as a substrate recruiting module for the DDB1–CUL4A–Roc1–RBX1 E3 ubiquitin ligase complex [12,13] and the immunomodulatory compounds thalidomide, lenalidomide, pomalidomide, and avadomide (Figure 1) alter its recruiting functions by attracting various “neo” protein substrates including Ikaros, Aiolos, [14] and casein kinase 1α (CK1α) [15] to the DDB1-CUL4A-Rbx1 complex (Figure 2). It is now well established that immunomodulatory drug-dependent degradation of Ikaros and Aiolos potentiates human T cells [16] and is responsible for the cytotoxicity of human MM cells [17]. CK1α degradation is selectively targeted by lenalidomide which garners an apoptotic phenotype that is uniquely associated with clinical responses in del(5q) MDS [18].
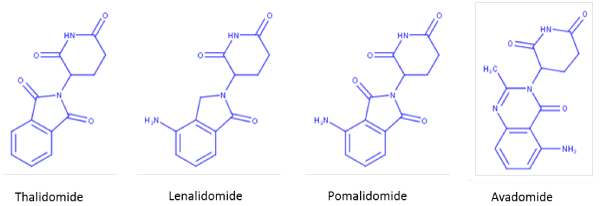
Figure 1. Structure of thalidomide and related immunomodulatory drug class.
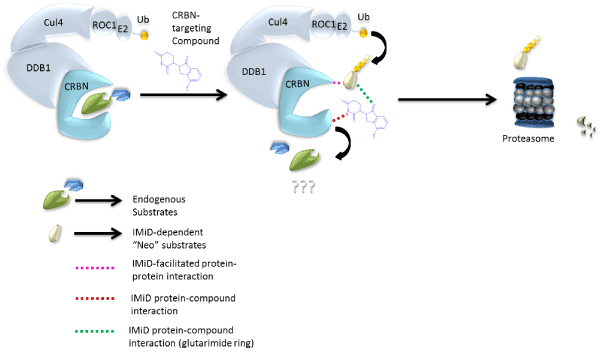
Figure 2. Model of immunomodulatory-drug mechanism of activation by binding Cereblon (CRBN).
Despite advances in our understanding of immunomodulatory drugs and their molecular mechanism, the notion that mice and other rodents are “resistant” to immunomodulatory compounds has been widely circulated due to the initial erroneous safety reports. In the thalidomide binding domain (TBD) of CRBN, examination of the sequences across vertebrate species revealed that CRBN is structurally well-conserved except for three amino acids (Cys366 human to Ser369 mouse, Glu377 to Val380 mouse, and Val388 human to Ile391 mouse) [19]. Interestingly, Ile391 is also present in chicken CRBN and in all non-mammal vertebrates excluding the flying lemur. Structural studies have now demonstrated how immunomodulatory drugs form a molecular bridge between CRBN and its drug-dependent binding partner CK1α [15]. Interestingly, Ile391 introduces a steric hindrance that diminishes the binding of all known neo-substrates that contain a conserved β-hairpin loop in their secondary structure including CK1α, Ikaros and Aiolos [15]. Explicitly, in the lenalidomide-CRBN-CK1α interaction, additional Van der Waals forces are formed at Val388, which stabilizes the complex and enhances the ubiquitin degradation of the substrates [15].
Consistent with the current premise on the role of Ile391, we confirmed that Ikaros ubiquitin-mediated degradation fails to occur in mouse T cells and multiple myeloma tumor cells [19]. We also demonstrated functional resistance of mouse T cells with regard to interleukin-2 production and proliferation. Mouse multiple myeloma cell lines showed no evidence of apoptosis in response to immunomodulatory drug treatment in comparison to potent suppression of several human multiple myeloma cell lines. CRBN protein expression levels were similar in most of the mouse and human cell lines tested which is consistent with the possibility that mouse cells are inherently resistant to immunomodulatory drugs and thus fundamentally different with regard to CRBN function. This is also consistent with the idea that Ikaros/CK1α recruitment is indeed responsible for T cell potentiation [15,19] and responsible for the cytotoxic effects of these drugs in human tumor cells [15,19].
Consequently, the negative impact of Ile391 in response to immunomodulatory drugs raises crucial questions about the physiological performance of CRBN in rodents and in other vertebrates possessing this amino acid variant. If non-drug-induced CRBN substrates (i.e., native substrates) contain a similar β-hairpin conserved structure present in CK1α, Ikaros and Aiolos, then Val388 may represent a functionally inactive variant selected through evolutionary adaptation in mammals. Using theoretical modeling studies and orthogonal drug binding assays to several CRBN TBD sequence variants, we demonstrated that the Ile391 CRBN TBD protein does not alter its binding affinity with immunomodulatory drugs [19] suggesting that this variant may retain at least some of the physiological properties.
Given the conserved immunomodulatory drug binding properties, we then hypothesized that target recruitment may be selectively compromised by Ile391 or other variants of CRBN. Since all known immunomodulatory drug-induced neo-substrate recruitment occurs through an interaction with a conserved β-hairpin-loop structure, we exploited a new technology called proteolysis targeting chimeras (PROTAC) to replace the substrate binding capacity of CRBN [20]. This new technology was first developed more than a decade ago as a means of selectively recognizing intracellular proteins and triggering their destruction for scientific and therapeutic applications. These compounds contain complementary bifunctional components that 1) interact with specific proteins, and 2) engage an E3 ligase to trigger the proteasome-mediated degradation of specified proteins of interest. Thalidomide–related compounds are key components of PROTAC technology due their fidelity in hijacking the CRBN/DDB1/Cul4A/Rbx1 E3 ubiquitin ligase complex (20). Using dBET1, a thalidomide-JQ1 PROTAC, that degrades BET domain proteins [21], we investigated the E3-ubiquitin ligase function of mouse CRBN. Interestingly, we demonstrated that mouse CRBN can bind thalidomide containing PROTACs with similar binding affinity, trigger CRBN-dependent degradation of BRD4, and thus confirmed that the CRBN/DDB1/Cul4A/Rbx1 complex is functional in mouse cells.
It is cogent to postulate that there is a resistance mechanism related to the V388I mutation and reduction in Ikaros, Aiolos, and CK1α degradation. However, chicken CRBN, and other vertebrate species, also exhibit this mutation, but show sensitivity to thalidomide. CRBN-dependent developmental defects after thalidomide treatment have been definitively shown in several Ile391-containing species [12]. Therefore, it is possible that teratogenicity, and perhaps other effects of immunomodulatory drugs, occur through a distinct mechanism. CRBN interacts intracellularly with calcium-activated potassium channels and the metabolic sensor adenosine monophosphate-activated protein kinase (AMPK), which in the latter, suppresses its activation and its downstream target mammalian target of rapamycin (mTOR) [22-24]. Collectively, this data suggests that there may be multiple endogenous substrates of CRBN and that the immunomodulatory drugs may influence its physiological function through different mechanisms. One study in particular proposes a ubiquitin-independent role for CRBN as a chaperon for the plasma membrane glycoprotein, CD147, and the monocarboxylate transporter, MCT1, that transports lactate [25]. In this study, researchers showed that the CD147-MCT1 complex is vital for the regulation of cellular metabolism and expression of fibroblast growth factor 8, which is the transcription factor reported to mediate thalidomide-associated teratogenicity in zebrafish and chickens [12]. Furthermore, through elegantly designed experiments, these investigators showed that thalidomide treatment destabilizes the CD147-MCT1 complex and that a morpholino-induced loss of CD147 suppresses limb development similar to thalidomide. Collectively, this study suggests that destabilization of the CD147-MCT1 complex is responsible for immunomodulatory drug-induced teratogenicity rather than degradation of the established neo-substrates [25].
Few medical disasters have reached the level of thalidomide-associated teratogenicity and had such prevailing consequences in its aftermath. Elucidation of thalidomide’s molecular target has been widely applauded as a major scientific breakthrough. Insights into aspects of CRBN biology relevant for the preclinical development of PROTAC compounds in humans are likely to continue for decades. However, we highlight the potential disadvantages of using CRBN-targeting PROTAC compounds as they all may harbor teratogenic potential, but should be tested in rodents and other relevant animal models during pre-clinical development. Using these new agents, it may be possible to appreciate the physiological roles for CRBN in protein regulation since it is evident that it plays a fundamental role in both proteasome and non-proteasome-related cellular regulation as well as in fetal development.
- Ward SP (1962) Thalidomide and congenital abnormalities. Br Med J 2: 646-647. [Crossref]
- Diggle GE (2001) Thalidomide: 40 years on. Int J Clin Pract 55: 627-631. [Crossref]
- Peltzman S (1973) An Evaluation of Consumer Protection Legislation: The 1962 Drug Amendments. J Polit Econ 81: 1049-1091.
- D'Amato RJ, Loughnan MS, Flynn E, Folkman J (1994) Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A 91: 4082-4085. [Crossref]
- Haslett PA, Corral LG, Albert M, Kaplan G (1998) Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med 187: 1885-1892. [Crossref]
- Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, et al. (1999) Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. N Engl J Med 341: 1565-1571. [Crossref]
- Thomas DA (2000) Pilot studies of thalidomide in acute myelogenous leukemia, myelodysplastic syndromes, and myeloproliferative disorders. Seminars in Hematology 37: 26-34.
- Zeldis JB, Williams BA, Thomas SD, Elsayed ME (1999) S.T.E.P.S.: a comprehensive program for controlling and monitoring access to thalidomide. Clin Therap 21: 319-330. [Crossref]
- Larsen V (1963) The Teratogenic Effects of Thalidomide, Imipramine Hc1 And Imipramine-N-Oxide Hc1 On White Danish Rabbits. Acta Pharmacol Toxicol (Copenh) 20: 186-200. [Crossref]
- Fickentscher K, Köhler F (1974) Teratogenicity and embryotoxicity of thalidomide and 3-aza-thalidomide in mice. Pharmacology 11: 193-198. [Crossref]
- Giroud A, Tuchmann-Duplessis H, Mercier-Parot L (1962) Observatios on the teratogenic repercussions of thalidomide in the mouse and rabbit. C R Seances Soc Biol Fil 156: 765-768.
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, et al. (2010) Identification of a primary target of thalidomide teratogenicity. Science 327: 1345-1350. [Crossref]
- Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, et al. (2014) Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512: 49-53. [Crossref]
- Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, et al. (2014) Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343: 301-305. [Crossref]
- Petzold G, Fischer ES, Thoma NH (2016) Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 532: 127-130. [Crossref]
- LeBlanc R, Hideshima T, Catley LP, Shringarpure R, Burger R, et al. (2004) Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood 103: 1787-1790. [Crossref]
- Quach H, Ritchie D, Stewart AK, Neeson P, Harrison S, et al. (2010) Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 24: 22-32. [Crossref]
- Krönke J, Fink EC (2015) Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523: 183-188. [Crossref]
- Akuffo AA, Alontaga AY, Metcalf R, Beatty MS, et al. (2018) Ligand-mediated protein degradation reveals functional conservation among sequence variants of the CUL4-type E3 ligase substrate receptor cereblon. J Biol Chem 293: 6187-6200. [Crossref]
- Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, et al. (2003) Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics 2: 1350-1358. [Crossref]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, et al. (2015) DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348: 1376-1381. [Crossref]
- Jo S, Lee KH, Song S, Jung YK, Park CS (2005) Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J Neurochem 94: 1212-1224. [Crossref]
- Kang JA, Park SH, Jeong SP, Han MH, Lee CR, et al. (2016) Epigenetic regulation of Kcna3-encoding Kv1.3 potassium channel by cereblon contributes to regulation of CD4(+) T-cell activation. Proc Natl Acad Sci U S A 113: 8771-8776. [Crossref]
- Lee KM, Jo S, Kim H, Lee J, Park CS (2011) Functional modulation of AMP-activated protein kinase by cereblon. Biochim Biophys Acta 1813: 448-455. [Crossref]
- Eichner R, Heider M, Fernández-Sáiz V, et al. (2016) Immunomodulatory drugs disrupt the cereblon-CD147-MCT1 axis to exert antitumor activity and teratogenicity. Nat Med 22: 735-743. [Crossref]