Cisplatin chemotherapy easily induces a resistant phenotype in tumor cells. In p53-deficient tumor cells, cisplatin causes G2 arrest, which allows cells to repair DNA damage. We attempted to visualize how cisplatin-induced G2 arrest is modified by Chk1 inhibitor or paclitaxel (PTX) and examine the effects on cell survival in both parental and cisplatin-resistant tumor cells. We used HeLa cells expressing fluorescent ubiquitination-based cell cycle indicator (Fucci). Cisplatin-resistant HeLa-Fucci (CR) cells were established by incubating parental HeLa cells with doses of cisplatin that increased in a stepwise manner. Time-lapse images were acquired by fluorescence microscopy, and surviving fractions were determined by colony-forming assay. Cisplatin treatment caused parental cells to accumulate green cells, representing G2 arrest, whereas combined therapy with a Chk1 inhibitor abrogated this effect, leading to a remarkable sensitization. CR cells did not exhibit G2 arrest, and Chk1 inhibitor did not influence the cell cycle, accompanied by slight reduction in survival. PTX arrested both cells, to similar extents, in mitosis with abnormal fluorescence; however, parental cells were arrested in G2 phase before entering mitosis, whereas CR cells were not, when cisplatin was used in combination. Moreover, cell survival increased in parent cells, but not in CR cells, relative to treatment with PTX alone. In conclusion, we should carefully consider G2 arrest kinetics, depending on the presence or absence of cisplatin resistance, in efforts to sensitize cells to cisplatin-based chemotherapy.
cisplatin, G2 arrest, DNA damage, cisplatin resistance, fluorescent ubiquitination-based cell cycle indicator (Fucci)
Cisplatin is a commonly used chemotherapeutic agent that forms monoadducts, and intra-strand cross-links and inter-strand cross-links with DNA [1]. Of these lesions, inter-strand crosslinks make the predominant contribution to cell death, even though their yield is quite low [1-3]. These lesions can activate cell-cycle checkpoints that mediate the cell-cycle arrest required for repair of DNA damage [4]. In particular, tumors with p53 dysfunction depend strongly on G2/M checkpoint activation [5,6]. Thus, abrogation of G2/M checkpoints could lead to sensitization of cisplatin-treated tumor cells with deficiency in p53; therefore, Chk1 and Wee1 inhibitors are being developed as sensitizers for DNA-damaging agents [6,7]. On the other hand, cisplatin induces a resistant phenotype in many types of tumor cells; the underlying mechanisms, which have been well studied, include increased drug efflux, enhanced detoxification, increased DNA repair, and alterations in oncogenes [8]. However, the differential effects of G2/M checkpoint inhibition on drug sensitivity of parental and cisplatin-resistant cells remain largely unclear. Furthermore, paclitaxel (PTX), an anti-microtubule agent, is often combined with cisplatin in clinical settings [9]. PTX agent arrests cells in mitosis [10]; therefore, complicated cell-cycle kinetics should be induced when PTX and cisplatin are combined.
Sakaue-Sawano et al. developed the Fucci methodology for visualizing cell-cycle progression using fluorescent proteins: Fucci-expressing cells in G1 and other phases emit red and green fluorescence, respectively [11], allowing us to detect changes in cell-cycle stage in live cells. Taking advantage of this system, we explored the effect of G2/M checkpoint modulation on G2 arrest and cell survival, using HeLa cells expressing the Fucci probes (HeLa-Fucci) and a cisplatin-resistant cell line derived from parental HeLa-Fucci cells (CR). For simplicity, we focused on conditions in which G2 arrest occurred only in parental cells following a certain cisplatin dose, and then explored the effects described above.
Cell lines and culture conditions
HeLa cells expressing the Fucci probes (HeLa-Fucci cells) were obtained from the RIKEN Bio-Resource Center through the National Bio-Resource Project of MEXT, Japan. Cells were maintained at 37°C in a 5% CO2 humidified atmosphere in DMEM (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum.
Establishment of cisplatin-resistant cell lines
HeLa-Fucci cells were seeded in 100mm dishes and sub-cultured in increasing concentrations of cisplatin (Sigma-Aldrich). Briefly, cells were first cultured in 0.1μM cisplatin and the medium containing cisplatin was replaced twice a week. Surviving cells were passaged once a week. After 7 treatments with 0.1μM cisplatin, the concentration was increased to 0.2μM, and the treatment was repeated twice. Finally, the concentration was increased to 0.4mM, and cells that survived more than 16 treatments were designated cisplatin-resistant (CR) cells. In general, CR cells treated with 0.4μM cisplatin around 50 times were used in the experiments.
Immunostaining for 53BP1
Cells grown on Lab-Tek Chamber slides (Nunc, Rochester, NY, USA) were treated with 20μM cisplatin for 1h. Thereafter, cells were incubated in the absence of cisplatin, and then fixed in 4% paraformaldehyde for 30 min at the indicated times after treatment. Fixed cells were then incubated in anti-53BP1 antibody (1:500) (Abcam, Cambridge, MA, USA) for 1h. After extensive washing in Tris-buffered saline plus Triton X-100(TBS-T), cells were incubated in anti-rabbit IgG conjugated with Alexa Fluor 647 (1:500) (Life Technologies, Carlsbad, CA, USA) for 30 min. Finally, slides were washed in TBS-T and mounted with ProLong Gold Antifade Reagent (Life Technologies) with DAPI. Fluorescence was observed using a BIOREVO BZ-9000 fluorescence microscope (KEYENCE, Osaka, Japan).
Drug treatment and time-lapse imaging
Cells grown on 35mm plastic cultured dishes were treated with 5μM cisplatin alone for 1h or 300nM Chk1 inhibitor (PF-00477736) (Sigma-Aldrich) for 24h in the presence of 5μM cisplatin for the first 1h. Cells were also treated with 20nM paclitaxel (PTX) (Wako Pure Chemical Industries, Osaka, Japan) for 24h in the presence or absence of 5μM cisplatin for the first 1h. Time-lapse imaging of drug-treated cells was taken using a BIOREVO BZ-9000 fluorescence microscope (KEYENCE) immediately after each treatment. For time-lapse imaging, cells were held in an incubation chamber at 37°C in a humidified atmosphere containing 5% CO2 (Tokai Hit, Fujinomiya, Japan). Images were acquired every 2h.
Colony-forming assay
After drug treatment, cells were trypsinized, and the appropriate numbers of cells were plated in 60 mm dishes. After incubation for 10–14 days, colonies were fixed and stained in crystal violet solution. Colonies consisting of more than 50 cells were counted, and survival fractions were calculated in triplicate.
Statistical analysis
Student’s t-test was used for statistical analysis. P values <0.05 were considered statistically significant.
Characterization of cisplatin-resistant cells
First, we established cisplatin-resistant HeLa-Fucci (CR) cells, which are able to grow in growth medium containing 0.4μM cisplatin, as described in Materials and Methods. Morphologies of parental and CR cells in exponentially growing phase are shown in Figure 1A. Parental and CR cells exhibited epithelial cobblestone-like (Figure 1Aa) and mesenchymal spindle-like (Figure 1Ab) morphologies. As expected, CR cells could grow in growth medium containing 0.4μM cisplatin, whereas parental cells could not (Figure 1B). Cisplatin resistance, as determined by colony-forming assay, was observed in cells treated 16 times with 0.4mM cisplatin; cells did not exhibit additional resistance after treatments were repeated more than 16 times (Figure 1C). We next examined the kinetics of DNA Double-Strand Breaks (DSBs) following cisplatin treatment (Figure 1D), which are formed in the course of inter-strand cross links repair [3]. 53BP1 was used as a marker of DSBs [12]. Both parental and CR cells exhibited small 53BP1 foci, representing DSBs, but fewer than 20 foci were present per cell. Thirty minutes after treatment, cells with more than 20 DSBs were detectable in parental cells, and the number increased for at least 24h after treatment. By contrast, in CR cells, the increment in foci number was first observed 24h after treatment. These results suggest that some mechanisms [8] might ultimately lead to lower induction of DSBs in CR cells, resulting in cisplatin resistance.
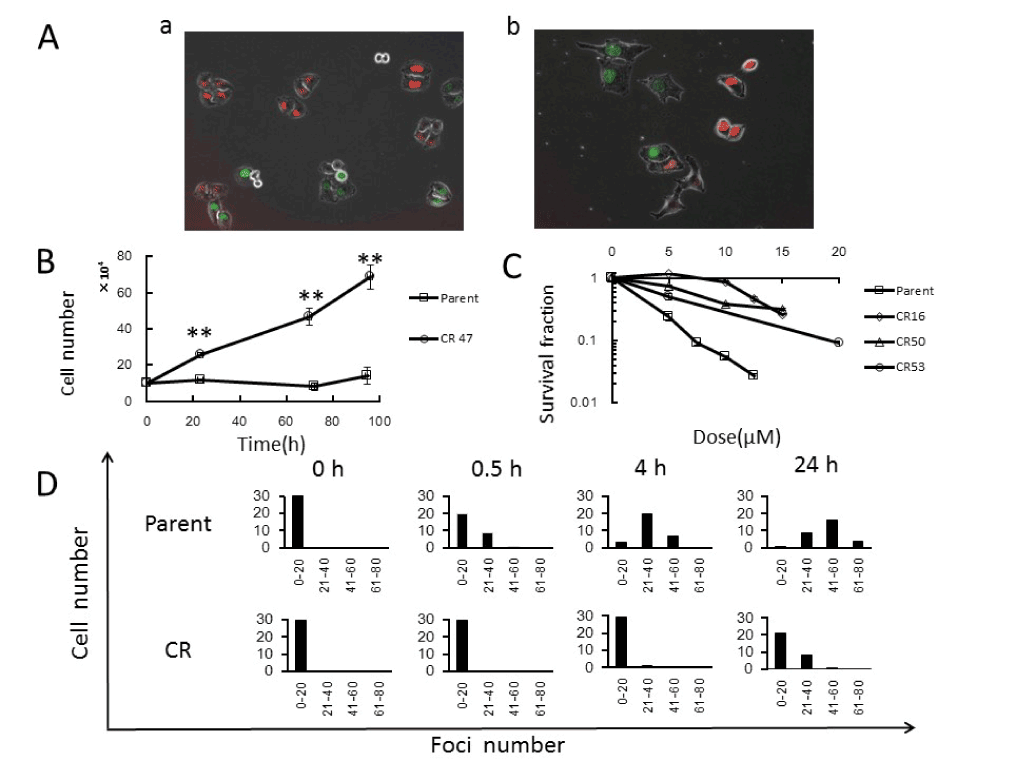
Figure 1. Characterization of cisplatin-resistant HeLa-Fucci (CR) cells
A. Morphologies of exponentially growing parental (a) and CR (b) cells. B. Growth curves in parental and CR cells in growth medium containing cisplatin. Parental and CR cells were counted in growth medium containing 0.4μM cisplatin. Data represent means S.D. of triplicate samples. C. Dose–cell survival curves in parent and CR cells following cisplatin treatment. CR16, CR50, and CR53 represent the number of treatments with 0.4μM cisplatin. Cells were treated with cisplatin at the indicated doses for 1h, and colony-forming assays were performed as described in Materials and Methods. Data represent means S.D. of triplicate samples. D. Kinetics of DNA double-strand breaks (DSBs) in parental and CR cells following cisplatin treatment. Cells were treated with 20μM cisplatin for 1h, and then immunostained for 53BP1 as a marker of DSBs at the indicated times after treatment. Histograms of fluorescence foci numbers are shown.
Fucci fluorescence kinetics following cisplatin or PTX treatment alone
As described in the Introduction, we chose a cisplatin treatment condition (5μM, 1h; based on Figure 1C) in which parental cells were arrested in G2 phase whereas CR cells continued to grow. After cisplatin treatment alone, we monitored the change in fluorescence (Figure 2). Parental cells exhibited a gradual increase in the number of green cells, representing G2 arrest, reaching a peak around 16 h after treatment. This observation was confirmed by flow-cytometric analysis of DNA contents (data not shown). On the other hand, CR cells did not exhibit significant accumulation of green cells (Figure 2A,2B).
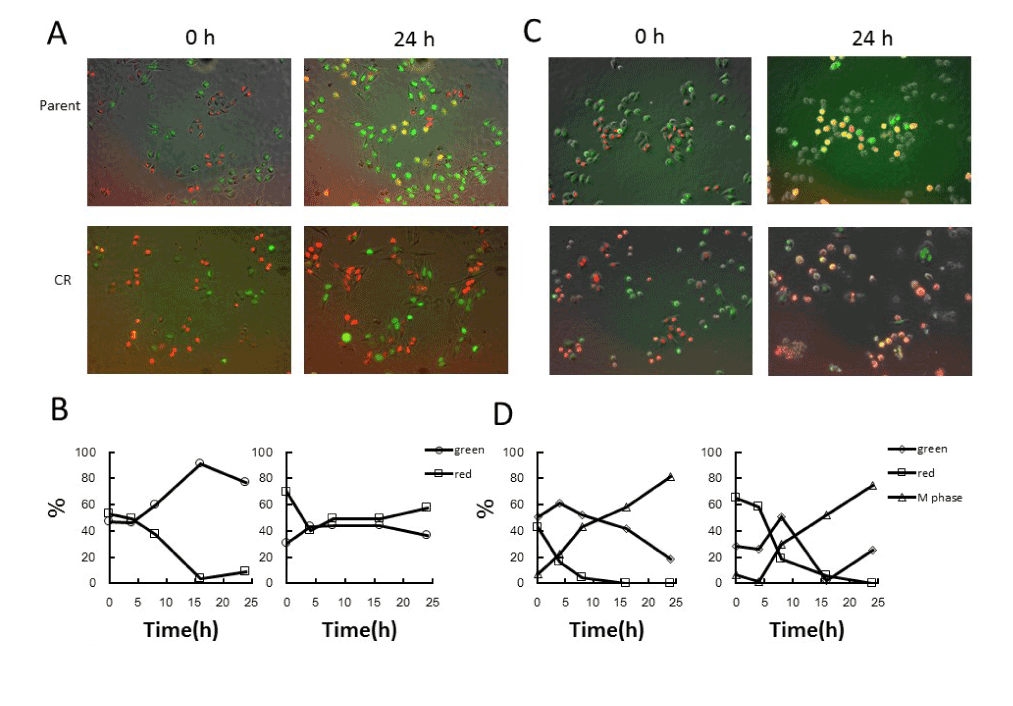
Figure 2. Fluorescence kinetics in parental and CR cells following cisplatin or PTX treatment alone
A. Fluorescence imaging in parental and CR cells following cisplatin treatment alone. Cells were treated with 5μM cisplatin for 1h, and fluorescence images were taken after treatment. B. Quantitative analysis of the proportions of red and green cells following treatment. The percentages of green and red cells were plotted as a function of times after treatment. Left panel, parent cells; Right panel, CR cells. C. Fluorescence imaging in parental and CR cells following PTX treatment alone. Cells were treated with 20nM PTX for 24h, and fluorescence images were taken during treatment. D. Quantitative analysis of the proportions of red (interphase), green (interphase), and mitotic cells during the treatment. The percentages of green (interphase), red (interphase), and mitotic cells were plotted as a function of times after treatment. Left panel, parental cells; right panel, CR cells.
PTX is an anti-microtubule agent that arrests growing cells in mitosis, leading to mitotic catastrophe [10]. PTX is often combined with cisplatin in clinical settings [9]. We previously reported that HeLa-Fucci cells produce abnormal fluorescence at mitosis after treatment with anti-microtubule agents: red fluorescence is also emitted, which never occurs under normal conditions when only green fluorescence is emitted [13]. When both types of cells were treated with PTX, we observed many mitotic round cells exhibiting abnormal fluorescence 24 h after the start of treatment (Figure 2C,2D).
The effect of Chk1 inhibitor on cisplatin-induced cell-cycle kinetics and cell survival
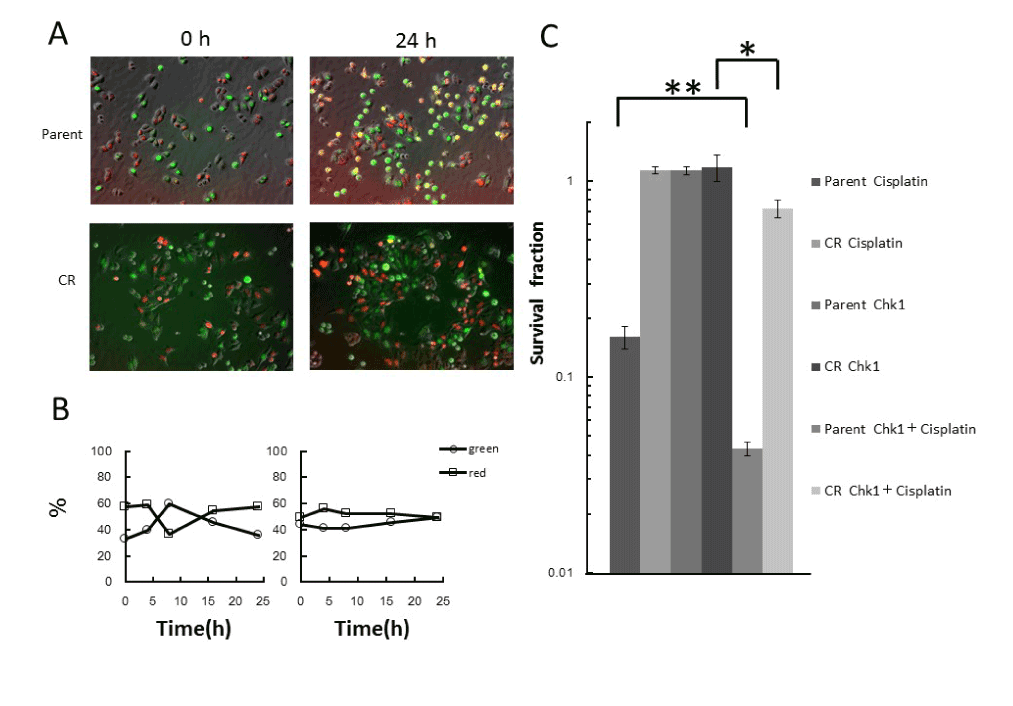
When Chk1 inhibitor, which is able to release G2 arrest, was combined with cisplatin treatment, no accumulation of green cells was detected in parental cells (Figure 3A,3B). Similarly, no change was detected in CR cells (Figure 3A,3B). A remarkable reduction in the surviving fraction was observed in parental cells, but not in CR cells. Chk1 inhibitor alone did not exert any effect on cell survival (Figure 3C).
2021 Copyright OAT. All rights reserv
Figure 3. Effect of Chk1 inhibitor in combination with cisplatin treatment
A. Fluorescence kinetics in parental and CR cells during treatment with cisplatin in combination with Chk1 inhibitor treatment. Cells were treated with 300nM Chk1 inhibitor for 24h in the presence of 5μM cisplatin for the first 1h, and fluorescence images were taken at the indicated times during the treatment. B. Quantitative analysis of the proportions of red and green cells during combined treatment. The percentages of green and red cells were plotted as a function of times during the treatment. Left panel, parent cells; right panel, CR cells. C. Surviving fractions, as determined by colony-forming assay under the same conditions used for time-lapse imaging. Data represent means S.D. of triplicate samples. *p<0.01, **p<0.001.
The effect of combined treatment with cisplatin and PTX on cell-cycle kinetics and cell survival
In parental cells, when cisplatin and PTX were combined, the number of green cells increased, and mitotic cells were rarely detectable 24h after treatment; by contrast, abnormal mitotic cells were observed in CR samples (Figure 4A,4B), similar to what was observed after treatment with PTX alone (Figure 3A,3B). Moreover, the surviving fraction in parental cells rose significantly relative to that in cells subjected to PTX treatment alone. After the same treatment, the surviving fraction of CR cells did not change (Figure 4C).
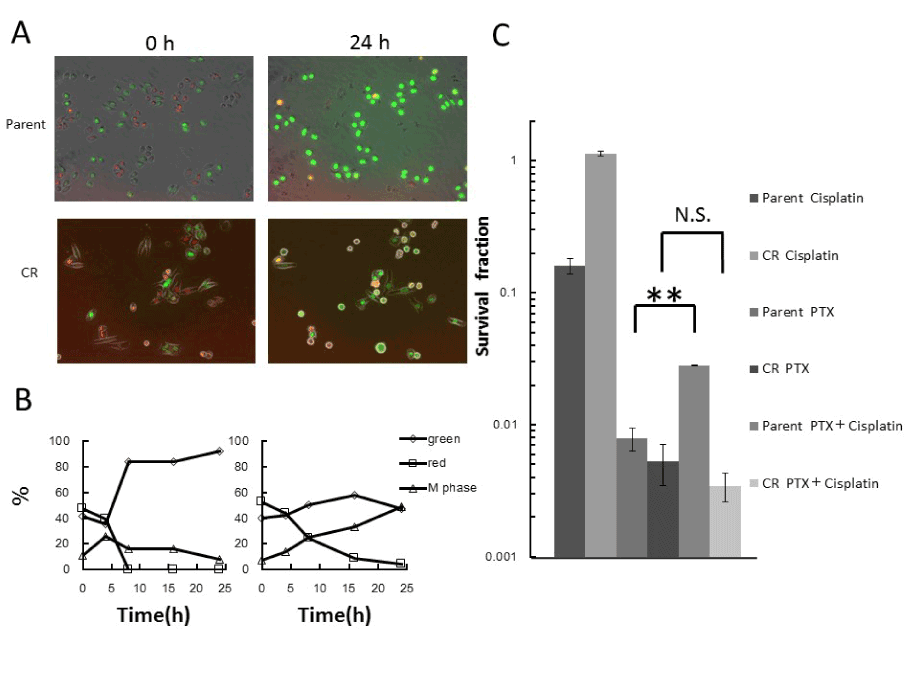
Figure 4. Effect of PTX treatment when combined with cisplatin treatment
A. Fluorescence kinetics in parental and CR cells during combined treatment with cisplatin and PTX. Cells were treated with 20nM PTX for 24h in the presence of 5μM cisplatin for the first 1h, and fluorescence images were taken at the indicated times during treatment. B. Quantitative analysis of the proportions of green (interphase), red (interphase), and mitotic cells during combined treatment. The percentages of green (interphase), red (interphase), and mitotic cells were plotted as a function of times during treatment. Left panel, parent cells; Right panel, CR cells. C. Surviving fractions, as determined by colony-forming assay under the same conditions used for time-lapse imaging. Data represent means S.D. of triplicate samples. **p<0.001; N.S: Not Significant.
Cisplatin is a widely used chemotherapeutic agent and relatively easily induces cisplatin resistance in varying tumors [8]. Currently, G2/M checkpoint inhibitors are being developed to sensitize tumor cells, especially those with deficient p53, and PTX is often combined with cisplatin in order to increase cellular sensitivity [9]. However, it remains unclear how G2/M checkpoint inhibitors or PTX influence cisplatin-sensitive and -resistant cells. In this study, we focused on conditions in which G2 arrest occurred only in sensitive cells following a certain cisplatin dose, and examined how combined treatment with Chk1 inhibitor or PTX affect cell-cycle kinetics and survival in cisplatin-sensitive and -resistant cells.
Previously, preparation of single-cell suspensions after fixation was required for flow-cytometric analysis to detect G2 arrest following exposure to DNA-damaging agents [14]. However, the Fucci system allows us to observe accumulation of green cells as a marker of G2 arrest in living cellsusing this system [11], we could easily detect cisplatin-induced G2 arrest in parental cells (Figure 2). In parental cells, abrogation of G2 arrest was also clearly observed when Chk1 inhibitor was administered in combination (Figure 2); by contrast, CR cells did not exhibit any changes. Cell survival assays revealed that G2 arrest contribute to survival of parental cells, but factors such as reduced incorporation of cisplatin to DNA or enhanced DNA repair [8] in CR cells might ultimately decrease DNA damage below the threshold level required to activate G2 arrest. The Fucci system also detected similar cell-cycle kinetics in both parental and CR cells following PTX treatment alone, accompanied by a similar level of cell survival (Figure 4). It was reasonable to predict that combined therapy with cisplatin would arrest the cell cycle at G2 phase only in parental cells: because PTX kills cells in mitosis [10], cells that stop at G2 phase were able to escape from cell death induced by PTX under the conditions we used. This might explain the increase in survival of parental cells. The cell-cycle kinetics in CR cells was not influenced by cisplatin treatment; therefore, the surviving fraction was the same as that following PTX treatment alone.
In conclusion, we were able to visualize G2 arrest kinetics in both parental and CR cells following treatment with cisplatin, either alone or in combination with Chk1 inhibitor or PTX. Furthermore, we demonstrated that G2/M checkpoint inhibitor does not necessarily influence survival in cisplatin-resistant cells; instead, cisplatin-induced G2 arrest might inhibit PTX-induced cell death in cisplatin-sensitive cells. Thus, cisplatin-induced G2 arrest kinetics should be considered carefully, depending on the presence or absence of cisplatin-resistance, in efforts to sensitize cells to cisplatin-based chemotherapy.
We thank Dr. A. Miyawaki and Dr. A. Sakaue-Sawano for providing HeLa cells expressing the Fucci probes.
This study was supported in part by Grants-in-Aid for Scientific Research from MEXT (26861569, 26293399, and 25670796) to A.K. and M.M.
- Kartalou M, Essigmann JM (2001) Recognition of cisplatin adducts by cellular proteins. Mutat Res 478: 1-21. [Crossref]
- Diggle CP, Bentley J, Knowles MA, Kiltie AE (2005) Inhibition of double-strand break non-homologous end-joining by cisplatin adducts in human cell extracts. Nucleic Acids Res 33: 2531-2539. [Crossref]
- Hlavin EM, Smeaton MB, Miller PS (2010) Initiation of DNA interstrand cross-link repair in mammalian cells. Environ Mol Mutagen 51: 604-624. [Crossref]
- Suganuma M, Kawabe T, Hori H, Funabiki T, Okamoto T (1999) Sensitization of cancer cells to DNA damage-induced cell death by specific cell cycle G2 checkpoint abrogation. Cancer Res 59: 5887-5891. [Crossref]
- Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, et al. (1996) UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst 88: 956-965. [Crossref]
- Osman AA, Monroe MM, Arves MVO, Patel AA, Katosonis P, et al. (2014) Wee-1 kinae inhibition overcomes ciplatin resistance associated with high risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther. [Epub ahead of print] [Crossref]
- Choudhury A, Cuddihy A, Bristow RG (2006) Radiation and new molecular agents part I: targeting ATM-ATR checkpoints, DNA repair, and the proteasome. Semin Radiat Oncol 16: 51-58. [Crossref]
- Lu HP, Chao CC (2012) Cancer cells acquire resistance to anticancer drugs: an update. Biomed J 35: 464-472. [Crossref]
- Behera M, Owonikoko TK, Kim S, Chen Z, Higgins K, et al. (2014) Concurrent therapy with taxane versus non-taxane containing regimens in locally advanced squamous cell carcinomas of the head and neck (SCCHN): a systematic review. Oral Oncol 50: 888-894. [Crossref]
- Fan W (1999) Possible mechanisms of paclitaxel-induced apoptosis. Biochem Pharmacol 57: 1215-1221. [Crossref]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, et al. (2008) Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132: 487-498. [Crossref]
- Panier S, Boulton SJ (2014) Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15: 7-18. [Crossref]
- Honda-Uezono A, Kaida A, Michi Y, Harada K, Hayashi Y, et al. (2012) Unusual expression of red fluorescence at M phase induced by anti-microtubule agents in HeLa cells expressing the fluorescent ubiquitination-based cell cycle indicator (Fucci). Biochem Biophys Res Commun 428: 224-229. [Crossref]
- Bernhard EJ, Maity A, Muschel RJ, McKenna WG (1995) Effects of ionizing radiation on cell cycle progression. A review. Radiat Environ Biophys 34: 79-83. [Crossref]