Electrolyte and renal abnormalities are an important but uncommon and under-appreciated secondary cause of cardiomyopathy (CM). Knowledge of these CMs is clinically relevant because they are potentially reversible following early diagnosis and prompt institution of appropriate treatment. However, electrolyte imbalance CMs has a paucity of research-based evidence to develop specific diagnostic and therapeutic guidelines while the few available clinical trials discuss the disease in the wider context of dilated cardiomyopathy. The evidence on the natural history, diagnosis and treatment of renal abnormalities (uraemic) CM are based on evidence from studies on CKD or ERSD patients yet these findings might not entirely apply to uraemic CM, which is one of the leading causes of mortality in CKD or ERSD patients. Furthermore, due to heterogeneous clinical presentation and non-pathognomonic features, diagnosis is often delayed, which is associated with an ominous prognosis. Knowledge of the natural history of electrolyte/renal abnormalities CMs is important to improve early diagnosis and the efficacy of treatment. Thus, this systematic review and meta-analysis aggregates published evidence on electrolyte imbalance and uraemic CMs to understand better their pathogenesis, manifestation, diagnosis and clinical management.
chronic kidney disease, end stage renal disease, electrolyte imbalance cardiomyopathy, hypocalcaemia, hypophosphatemia, uraemic cardiomyopathy
Electrolyte imbalance and renal abnormalities (in patients with chronic kidney disease [CKD] or end stage renal disease [ERSD]) are uncommon and under-appreciated secondary causes of cardiomyopathy (CM) – a heart muscle disease. The relationship between electrolyte imbalance and heart (HF) is long-established. Electrolyte imbalance is a frequent and potentially hazardous complication in heart failure (HF) patients. It arise due to pathophysiological alterations in the HF state leading to neurohormonal activation (stimulation of the renin-angiotensin-aldosterone system [RAAS] and sympathoadrenergic stimulation) and due to complications of standardized HF therapy – diuretics, cardiac glycoside or angiotensin converting enzymes – inhibitors (ACE-I) [1-3]. Increased knowledge of electrolyte imbalance in HF has contributed to increased therapeutic efficacy in HF patients but has also engendered new problems and complications related to a more vigorous therapy to warrant physicians treating HF patients to be cognizant of these advantages and dangers [1].
More recently, electrolyte imbalance have been implicated as an aetiology of CM, which if not treated might progress into HF. Indeed, the recent position and scientific statements by the European Society of Cardiology (ESC) [4,5] and the American Heart Association (AHA) [6,7] list electrolyte imbalance (calcium and phosphate) as one of the causes of secondary (non-genetic) CMs. However, published evidence from clinical trials on this type of CM are sporadic and a greater proportion of literature comes from case reports, whose evidence is less reliable relative to those based on clinical trials. The lack of data undermines a holistic understanding of the role of electrolyte imbalance in the pathogenesis of CM yet a good understanding of this CM is clinically essential because the disease is potentially reversible with appropriate therapy. On the other hand, cardiovascular (CV) complications especially uraemic CM is a leading cause of mortality in CKD or ESRD patients. Early diagnosis remains a clinical challenge since it presents without signs and symptom of HF with preserved systolic function. Further, clinical management of HF in uraemic patients remains unclear since current guidelines may not apply to patients with severe renal function. Thus, this paper performs a systematic review and meta-analysis of published evidence on CMs in the setting of electrolyte imbalance and renal abnormalities to improve the understanding on their prevalence, pathophysiology, clinical presentation, diagnosis and management.
Electrolyte is an umbrella term for electrically charged minerals and compounds occurring in blood urine, tissues, muscles and other body fluids, and are involved in many essential processes in the human body. Common electrolytes found in the human body include sodium, potassium, chloride, calcium, magnesium, phosphate and bicarbonate. Electrolyte balance is a key factor in maintaining homeostasis in the body as well as playing important roles in protecting cellular function, tissue perfusion, energy production, muscle contraction and acid-base balance [8]. Electrolyte imbalance is a common finding in many diseases [9,10]. The most important and prevailing electrolyte imbalances include hypo- and hyper-states of sodium, potassium, calcium and magnesium (Table 1).
Table 1. Common electrolytes, normal ranges and diseases
Electrolyte |
Normal Ranges |
Disease in the case of Imbalance |
Sodium Na+ |
135-145 |
Hyponatremia, Hypernatremia |
Chloride Cl- |
96-106 |
Hypochloremia, Hyperchloremia |
Potassium K+ |
3.5-5.5 |
Hypokalaemia, hyperkalaemia |
Magnesium Mg+2 |
1.7-2.2 |
Hypomagnesia, hypermagnesia |
Calcium Ca+2 |
8.5-10.2 |
Hypocalcaemia, hypercalcaemia |
Phosphorous PO4- |
2.5-4.5 |
Hypophosphatemia, hyperphosphatemia |
The kidney is the principally responsible organ for the retention and excretion of electrolytes in healthy individuals [11] although other mechanisms like hormonal interactions of antidiuretic hormone, aldosterone, and parathyroid hormone and other factors such as physiological stress also play important roles in regulating electrolyte balance [8]. Electrolyte imbalance are frequent in the elderly and critically ill patients and often occur in the progression of diseases such as diabetes mellitus, acute or chronic renal failure, severe cardiovascular (CV) events [12-14]. In children, electrolyte imbalance are common due to their small size, large ratio of surface area to volume and immature homeostatic mechanisms [8].
Electrolyte imbalance CM is an acquired (secondary) form of CM characterized by poor myocardial contractility and a dilated heart in the absence of known causes such as cardiac ischemia, hypertension, rheumatic heart diseases and congenital abnormalities as well as reversible causes such as alcohol, toxins, infection and metabolic abnormalities. Electrolyte imbalance may lead to many different diseases but those frequently associated with CM are hypo-states of calcium (hypocalcaemia) and phosphorous (hypophosphatemia) [4-7].
Overview
Hypocalcaemia is an electrolyte imbalance commonly encountered on routine clinical practice defined as defined as serum calcium levels less than 8.2 mg/dl. The condition may be transient, reversing with addressing the underlying cause expeditiously or chronic or even lifelong, when it is secondary to a genetic disorder or the result of irreversible post-surgical injury to the parathyroid gland or secondary to autoimmune destruction [14,15]. Hypocalcaemia is well known but a rare cause of DCM and should be considered in every patients with DCM because calcium supplementation reverses the disease [15]. The true prevalence of hypocalcemic CM remains unknown although epidemiological data of hypocalcaemia may provide a valuable insight into its prevalence. In the 1960s, hypocalcaemia had a prevalence of 18% in all hospitalized patients in primary and secondary care, and up to 85% in the intensive care unit [16]. In a more recent retrospective study based on laboratory results of 12,334 hospitalized patients performed between 2011 and 2014, hypocalcaemia accounted for 27.72% with a slight majority being male (57.07%). The highest prevalence was in patients older than 65 years (61.31%). There was a tendency towards decreasing incidence of hypocalcaemia from 35.42% in 2011 to 21.93 in 2014 [17]. In a five-year retrospective review of medical records of 3,989 patients between 2010 and 2015 in a Nigerian hospital, the prevalence of hypocalcaemia in pregnancy and puerperium (based on carpopedal spasms) was 1.3% [18].
Aetiology
The human body regulates serum calcium balance within the narrow range of 2.1 to 2.6 mmol/L by three main calcium-regulating hormones: parathyroid hormone (PTH), vitamin D, and calcitonin via their specific effects on the bowel, kidneys and skeleton [15]. PTH stimulates calcium absorption in the kidney and calcium release from bone as well as stimulates renal production of 1,25-dihydroxyvitamin D (calcitriol) from 25-hydroxyvitamin D. 1,25-dihydroxyvitamin D is the most active form of vitamin D and it acts on the gastrointestinal tract to increase calcium absorption. Humans obtain Vitamin D mainly through synthesis in the skin and diet. Skin synthesis requires exposure to ultraviolet light [15]. Thus, common causes of hypocalcaemia are thus vitamin D deficiency and hypoparathyroidism. Less common causes include resistance to PTH, vitamin D resistance, autosomal dominant hypocalcaemia, hypomagnesaemia and sclerotic metastases [16]. Other rare causes include hungry bone syndrome following parathyroidectomy for hyperparathyroidism, infusion of phosphate or calcium chelators such as citrate with massive blood transfusion, critical illness and after intravenous treatment with bisphosphates, particularly high-dose treatment in vitamin D deficient patients [15,16].
Vitamin D deficiency is a leading cause of hypocalcaemic CM. The presence of 1,25-dihydroxyvitamin D enhances intestinal absorption of calcium and phosphorus as well as promotes bone remodelling [15]. Vitamin D inadequacy (25-hydroxyvitamin D) concentration less than 75nmol/L is common in adults and children, which leads to reduced gastrointestinal calcium absorption of up to 50%, resulting in only 15% of dietary intestinal calcium absorbed [19]. Hypocalcaemia associated with vitamin D deficiency can occur in individuals whose exposure to ultraviolet light is low, particularly those with a diet low or missing in vitamin D. It may also occur during pregnancy and puerperium when Vitamin D requirements increase leading to low maternal vitamin D levels and as a result may cause hypocalcaemia in breastfed children [20]. Hypocalcaemia due to vitamin D deficiency manifests in some patients on antiepileptic medication that induce enzymes increasing vitamin D metabolism. The prevalence of vitamin D deficiency in institutionalized children with poor control of epilepsy is 75% [21]. Poor nutrition and minimal exposure to the sun possibly contributes to osteomalacia. Patients with small intestine disease such as coeliac disease that reduces dietary calcium and vitamin D absorption, may also exhibit hypocalcaemia. Less often, hypocalcaemia may manifest in individuals with profoundly low dietary intake of calcium but with vitamin D concentrations within the normal range [22].
PTH is another leading cause of hypocalcaemic CM. Hypocalcaemia occurs in patients with impaired function of the parathyroid glands, which is very common following thyroid or parathyroid surgery although it can also be idiopathic as seen mostly in young adults and less often as part of a genetic syndrome such autoimmune polyglandular syndrome type 1 [23]. Hypoparathyroidism is most common following inadvertent removal of or damage to the parathyroid glands or their vascular supply during total thyroidectomy in about 0.5% to 6% of total thyroidectomies [15]. Decreased levels of PTH results in excessive renal calcium loss and reduced intestinal absorption of calcium secondary to decreased production of 1,25- dihydroxyvitamin D. Sometimes, tissue resistance to PTH can produce biochemical pattern mimicking hypoparathyroidism, even though levels of the hormone are high. Such pseudo-hypoparathyroidism is due to the failure of PTH to activate its signalling pathway [24]. Pseudo-hypoparathyroidism is a genetically heterogeneous condition in which some patients may present with skeletal abnormalities such as Albright’s hereditary osteodystrophy, which can occur in other family members independent of any abnormality of serum calcium. Genetic mutations involving the development of parathyroid glands and synthesis or secretion of PTH can also lead to hypoparathyroidism. Genetic mutations can also lead to resistance to PTH at the proximal renal tubule, resulting in excessive renal calcium excretion and hypocalcaemia [15]. Severe hypocalcaemia can occur in patients with pre-existing vitamin D deficiency who receive intravenous bisphosphates because these compounds block bone resorption [25].
In addition to vitamin D deficiency and PTH, other causes are congenital abnormalities. Abnormalities in the embryonic development of the parathyroid glands may lead to DiGeorge syndrome associated with cardiac defects, abnormal facies, thymic hypoplasia, cleft palate and hypocalcaemia. Other genetic abnormalities capable of causing hypoparathyroidism or pseudo-parathyroidism [15]. Uncommon causes of hypoparathyroidism include heavy metal infiltration of the parathyroid glands with iron as seen in haemochromatosis or thalassemia. Copper deposition as seen in Wilson disease is also an uncommon cause, as it is metastatic infiltration of the parathyroid glands. Magnesium deficiency or excess can cause an impairment of PTH secretion and result in hypoparathyroidism [26].
Pathophysiology
Although the physiological role of serum calcium in the excitation and contraction of cardiac muscle fibres is well established but pathophysiology of hypocalcaemic CM remains unclear. During membrane depolarization, influx of calcium into the cardiomyocytes via voltage-gated L-type calcium channels raising the levels of cytosolic calcium available to bind troponin C [27-29]. This triggers the release of calcium from sarcoplasmic reticulum and subsequent binding of calcium ions to troponin-tropomyosin complex to form cross-bridges between actin and myosin filament resulting in muscle contraction [28,30,31]. Experimental animal models reveal reduced cardiac contractility in hypocalcaemia [32].
More recent evidence suggests that vitamin D and PTH may play an independent role in the pathogenesis of hypocalcaemic CM. There is increasing recognition of autocrine functions of vitamin D in several organs including cardiomyocytes. In experimental animal model, the ablation of vitamin D receptor in mice and vitamin D deficiency results in cardiac hypertrophy and fibrosis, which persists even after normalization of calcium levels with high-calcium and high-phosphate diet, suggesting vitamin D has an independent role in the causation of cardiac manifestations [33]. In other experimental models, vitamin D deficit in mothers resulted in delayed maturation and abnormal growth of cardiomyocytes in the offspring despite unchanged serum calcium concentration [34]. Epidemiological studies also report an association between deficits in vitamin D and HF. A community-based study enrolling elderly patients associated higher circulating vitamin D levels with better systolic function at baseline even after adjustment of CV risk factors and calcium, phosphate and PTH concentrations [35]. A related study of patients undergoing coronary angiography associated low levels of circulating vitamin D with depressed ejection fraction and increased CV mortality [36]. PTH plays an important role in the regulation of normal cardiac contractile function. PTH acts on voltage-gated calcium channel to exert a positive chronotropic effect in neonatal cardiomyocytes as well as stimulates the synthesis of intracellular proteins via stimulation of protein kinase C [37]. Case reports have also associated isolated hypoparathyroidism with significant reversal of LV systolic function [38,39].
Although pathogenic associations between myocardial contractility and calcium homeostasis have been generalised, neonates and adults show different mechanisms and hypocalcaemic cardiac manifestations. Vitamin D deficiency is common and a cause of hypocalcaemic CM in neonates [40-46]. Maternal deficits in vitamin D is the usual cause of vitamin D deficiency in neonates indicated by 96% of vitamin D deficient pregnant women positively correlating with low vitamin D levels in their infants [47]. On the other hand, in adults, vitamin D deficiency may be prevalent but significant LV systolic dysfunction with HF is uncommon with only 27 adult cases reported in literature [31] suggesting adult hypocalcaemic CM is not a manifestation of vitamin D deficiency in adults. In paediatric patients with hypocalcaemic CM, vitamin D deficiency is severe and associated with secondary hyperparathyroidism or due to relatively immature PTH response to hypocalcaemia or very high levels of PTH [45]. On the other hand, adult patients with hypocalcaemic CM hypoparathyroidism is common and usually the dominant metabolic abnormality and the presence of vitamin D deficiency may aggravate hypocalcaemia and LV systolic function [31]. However, there is no evidence that vitamin D supplementation may improve cardiac function in patients with hypocalcaemic CM although recent data suggests a tendency towards potential benefits [48,49]. The difference in age-related maturation of calcium handling mechanisms may explain differential effect of vitamin D deficits on serum calcium concertation and LV systolic function in neonates and in adults [31].
Clinical presentation
Clinical presentation of hypocalcaemic CM is not well described with current evidence derived from case reports, which reveal inconsistent findings suggesting that signs and symptoms are non-specific. Common signs and symptoms reported in case reports include complaints of weakness, progressive dyspnoea, sudden onset of chest heaviness and palpitations associated with tingling sensation in hand and arm for one month [31]. It is essential to ask about family history to identify a genetic cause or previous thyroidectomy. Patients who had undergone thyroidectomy may present with worsening breathlessness and orthopnoea [28]. In adults, worsening exertional dyspnoea associated with orthopnoea, but in the absence of paroxysmal nocturnal dyspnoea, chest pain, palpitations, syncope, cough, expectoration or febrile illness may suggest the need for tests for hypocalcaemia [27]. In elderly hypocalcemic CM patients, examination may reveal cardiac and non-cardiac signs and symptoms including mental confusion, psychomotor slowing, depressed facial expression, limb tremor, muscle spasms and crackling rales on lower half of both lungs [27]. Due to non-specific signs and symptoms, hypocalcaemic CM can easily be overlooked because the clinical symptoms of hypocalcaemia such as fatigue and muscle weakness are often regarded as subjective and ambiguous in the absence of tetany (a condition marked by intermittent muscular spasms due to malfunction of the parathyroid glands and a consequent of calcium deficiency) [29].
Clinical evaluation
Clinical evaluation for hypocalcaemic CM is a combination of laboratory investigations and non-invasive cardiac imaging. Initial diagnostic laboratory investigations include measurement for serum calcium, serum phosphorus, serum magnesium, serum 25-hydroxyvitamin D levels and serum PTH level. Hypocalcaemic patients will have low serum calcium corrected for albumin), low or inappropriately normal PTH levels, hyperphosphatemia, hypercalciuria and low 1,25-dihydrocyvitamin D levels [15]. ECG may reveal sinus tachycardia, T-wave inversion with a slightly increased corrected QT interval [28,30]. Chest radiography may demonstrate cardiac enlargement with pulmonary congestion and pleural effusion [28]. M-mode and 2D echocardiography reveal normal or enlarged chambers and severe generalized hypokinesia of LV with severely depressed ejected fraction with no evidence of wall abnormalities, transmitral flow with a restrictive physiology and preserved RV systolic function [28]. Troponin I may be elevated but BNP is within normal levels [30]. Cardiac MRI may be performed to clarify the clinical picture with morphological or functional alterations or contrast uptake in delayed enhancement sequences but its use is not well-defined [30].
Clinical management
Clinical management of hypocalcaemic CM targets treating the underlying condition and the management of signs and symptoms of HF. Calcium supplementation is the mainstay therapy for hypocalcaemic CM and is associated with improvement in cardiac structure and function [28,30,31]. If intravenous infusions are contemplated, hospitalization in an intensive care unit or specialized unit with access to cardiac monitoring and rapid ionized calcium determinations is ideal for optimal management and safety. Cardiac monitoring usually with ECG is recommended since dysrhythmias may occur if correction is too rapid [15,50]. Calcium gluconate is the preferred form of intravenous calcium because calcium chloride is more likely to cause local irritation. Treatment can be repeated until symptoms have cleared but this may only offer a temporary relief and thus continuous infusion of a dilute solution of calcium may be necessary to prevent recurrence of hypocalcaemia [16]. In patients on digoxin, calcium administration should be cautious with careful ECG monitoring because of increased sensitivity to fluctuations in serum calcium. Patients with hypocalcaemia associated with hypomagnesaemia may require magnesium supplementation therapy [16]. In patients with persistent (chronic) hypocalcaemia, supplementary treatment usually depends on the underlying hypocalcaemic cause [15]. Patients with vitamin D deficiency may require lower doses of oral vitamin D supplementation therapy either ergocalciferol (vitamin D2) or cholecalciferol (vitamin D3) [50]. However, patients with hypocalcaemic CM due to hypoparathyroidism may require higher and increasing doses of vitamin D (either calcitriol or alfacalcidol) because PTH is needed for the conversion of vitamin D to 1,25-dihydroxyvitamin D [51,52]. Patients with signs and symptoms of HF may receive standard HF therapy as per the current guidelines [30,31].
Overview
Phosphorous occurs throughout the body in skeleton, skeletal muscles and soft tissues and plays essential roles in cell structure (cell membrane and nucleic acids), cellular metabolism (generation of ATP), regulation of subcellular processes (phosphorylation od key enzymes) and the maintenance of acid-base homeostasis (urinary buffering) [53,54]. Phosphorous also forms the main structural element of membranes and bones. Intracellularly, phosphorous exist in the form of creatine phosphate, adenosine monophosphates and tripShosphates within bones and teeth (85%), soft tissues (14%) and extracellular fluids (1%) [55]. Normal serum concentration of phosphate is 1.7 to 4.5 mg/dL in adults and 4.0 to 7.0 mg/dL in children. In most laboratories reports, 10% of the inorganic orthophosphate form is bound to protein, 5% occurs in complex form with calcium and magnesium and 85% in the form of H2PO4- and HPO4-. [55]. The body maintains phosphate homeostasis through a balance of oral intake, gut absorption, kidney excretion and intracellular shifts. In clinical setting, low serum phosphorus levels are common with refeeding syndrome, continuous insulin treatment for diabetic ketoacidosis, sepsis, alcoholism, secondary hyperparathyroidism and tum-r-related osteomalacia. Congenital causes include X-linked hypophosphatemia and autosomal dominant hypophosphatemic rickets [54,55]. Severe Hypophosphatemic is defined by serum levels < 1.0 mg/dL (0.32 mmol/L) and may lead to ventricular arrhythmias, altered cardiac and respiratory functions and possibly cardiac death. The exact prevalence of hypophosphatemia is unknown but it is estimated at < 1% [54].
Aetiology
The general causes of hypophosphatemia in hospitalized patients include (i) decreased intestinal absorption of phosphorous; (ii) redistribution of phosphorous from the extracellular to the intracellular compartment; and (iii) increased loss of phosphorous through kidney or any combination of these processes [53-56]. Decreased dietary intake alone rarely causes hypophosphatemia probably because of enhancement of renal phosphate reabsorption to compensate for decreased intake [53]. Since vitamin D (1,25-dihydroxyvitamin D) is necessary for intestinal absorption of phosphate, its deficiency associated with the lack of exposure to sunlight or inadequate amounts in diet leading to intestinal malabsorption of vitamin phosphate. Liver disease associated with biliary obstruction may result in decreased production of 1,25-dihydroxyvitamin D [56]. In humans, phosphate-binding antacids can cause hypophosphatemia by preventing intestinal phosphate absorption [57-59]. Although significant hypophosphatemia does not occur when antacids are consumed with a normal diet, when phosphorous-deficient diet or in the setting of decreased phosphate concentrations, the use of phosphate-binding antacids may cause severe hypophosphatemia [53].
Increased renal excretion of phosphate is another common cause of hypophosphatemia. Increased urinary excretion of phosphate occurs in patients with primary hyperparathyroidism, and in those with secondary hyperparathyroidism associated with hypocalcaemia and intact renal function [54]. Renal tubular defects such as Fanconi syndrome and hereditary vitamin D-resistant rickets in humans can lead to phosphate wasting and moderate hypophosphatemia [53]. Diuretics such as acetazolamide, furosemide and thiazides mal also result in hypophosphatemia secondary to renal phosphate loss. Sodium bicarbonate administration can cause increased urinary phosphate excretion through increased fractional excretion of phosphate into the urine secondary to volume expansion and decreased ionized calcium secondary to alkalosis and dilution stimulates PTH release with subsequent phosphaturia. Chronic administration of glucocorticoids may cause hypophosphatemia by redistributing phosphate from extracellular fluid space to the intracellular space by increasing phosphate excretion in the urine [53,54].
A shift of phosphate out of the extracellular fluid into the intracellular space is the most common cause of hypophosphatemia in humans. An increase in the use of intracellular inorganic phosphate in glycolysis may be responsible for the intracellular shift of phosphate [60]. Conditions associated with an intracellular shift of phosphate include infusions of insulin or glucose, respiratory alkalosis and hyperalimentatin [60,61]. Recently, acute respiratory alkalosis and refeeding of malnourished patients (metabolic alkalosis) is a common cause of phosphorous redistribution with a greater reduction of phosphate observed in respiratory alkalosis [62,63]. During respiratory alkalosis, intracellular CO2 decreases causing intracellular pH to rise and stimulates the glycolytic pathway, enhances the production of sugar phosphates, which includes intracellular phosphorous entry and consequently decreasing serum phosphorous concentration [64]. Respiratory alkalosis also increases phosphorous uptake by muscles. The kidney response by increasing phosphate reabsorption independent of serum levels of phosphorous and becomes refractory to phosphaturic effect of PTH [54]. Finally, phosphorous redistribution of phosphorus and hypophosphatemia occur in leukaemia patients with rapidly replicating tumour cells, following hematopoietic reconstitution after allogenic peripheral blood stem cell transplantation and after parathyroidectomy [54].
Pathogenesis
The pathogenesis of hypophosphatemic CM is remains unclear. However, a systematic review of 11 primary studies comprising case reports or series, case-control study and animal studies performed between 1971 and 2015 suggested a number of possible pathogenic mechanisms [55]. The first is altered cardiac energy production. Phosphorous is involved in the glycolytic pathway in the generation of the high-energy phosphate bonds of adenosine triphosphate (ATP) and the formation of 2,3-diphosphoglycerate (2,3-DPG). The mechanisms of hypophosphatemic CM may be due to the depletion of ATP in cardiomyocytes and decreased 2,3-DPG in erythrocytes. ATP is the standard molecule for cellular energy in the human body. The heart has a high-energy demand with little reserves and thus cardiac demand for ATP is significant, constituting > 5 kg of ATP per day or 2 metric tons per year [65]. To meet this high cardiac energy demand, mitochondria occupies about 25% to 33% of each ventricular cell, whose primary function is to generate ATP to maintain cardiac contractility. Theoretically, the depletion of ATP can lead to cardiac contractile dysfunction. The use of 31P-magnetic resonance spectroscopy can measure cardiac ATP by providing an index of energetic state of the heart by mean of phosphocreatine-to-ATP ratio. However, the use of magnetic spectroscopy is limited to research due to low temporal and spatial resolution and low reproducibility [65]. Thus, limited use of magnetic spectroscopy in clinical setting denies evidence-based data to verify the altered cardiac energy theory of the pathogenesis of hypophosphatemic CM.
The second possible pathogenic mechanism of hypophosphatemic CM is impaired oxygen delivery to the tissues due to decreased production of ATP and 2,3-DPG. Hypophosphatemia due to parenteral nutrition has been shown to lead to a 52% reduction of ATP and 45% reduction of 2,3-DPG levels within erythrocytes. ATP and 2,3-DOG bind to haemoglobin and decrease cells’ affinity for oxygen resulting into its release into the tissues [66]. Normally, erythrocytes have nearly three-times more 2,3-DPG than ATP. Decreased intracellular 2,3-DPG results in a leftward shift of the oxy-haemoglobin dissociation curve and consequently tissue hypoxia. Under anaerobic conditions due to decreased tissue oxygen supply, one mole of glucose yield only two moles of ATP while under fully oxidized conditions, the same amount yields about 26 moles of ATP. Patients with severe HF have substantially reduced oxygen-haemoglobin binding and higher than normal 2,3-DPG levels, though to be an important mechanism to maintain adequate oxygen transport [67]. Eliminating this adaptive mechanism causes an increase in cardiac output and coronary blood flow to meet the same resting metabolic demands. In the setting of limited cardiac function and impaired coronary blood flow, a decrease in 2,3-DPG may potentially aggravate the imbalance between metabolic demands and tissue oxygen supple and lead to myocardial ischemia [55].
The third possible pathogenic mechanism of hypophosphatemic CM is the involvement of a recently identified molecule of the fibroblast growth factor (FGF) family, (FGF-23) – a serum constituent involved in phosphate metabolism. Alongside PTH and vitamin D, FGF-23 mobilizes sodium phosphate co-transporters in coordination with Klotho – a transmembrane protein with anti-aging properties. Patients with severe renal disease have high levels of circulating FGF-23 and are at risk for cardiac complications, suggesting the involvement of FGF-23 in cardiac disease. Moreover, in a study of 100 coronary care unit patients with renal insufficiency, Shibata et al. [68] positively correlated circulating FGF-23 levels with LV mass and reduced LV ejection fraction (LVEF), which is independent of renal function and other parameters related to calcium-phosphate metabolism. In contrast, in patients with chronic kidney disease, increased levels of FGF-23 is associated with hyperphosphatemia rather than hypophosphatemia suggesting that higher FGF-23 is just a bystander in patients with advanced kidney disease and cardiac diseases [68]. Such conflicting findings warrants further longitudinal studies to clarify the role of FGF-23 in cardiac disease.
Clinical presentation
Clinical presentation of patients with hypophosphatemic CM depends on serum phosphate levels, which varies from asymptomatic to severe symptoms including cardiac arrest [69-72]. Signs and symptoms of hypophosphatemic CM include muscle weakness, cardiac arrhythmias, rhabdomyolysis, paraesthesia, motor neuropathy, ataxia, hallucination, seizure, haemolysis, and insulin resistance [73]. Serious symptoms such as muscle weakness and cardiac arrhythmias often manifests when serum phosphate levels decrease to < 1.0 mg/dL [74]. Patients with severe hypophosphatemia have a greater incidence of ventricular arrhythmias in acute myocardial infraction and greater need for vasoactive drug titration after cardiac surgeries [75,76]. Correction of hypophosphatemia often leads to a reversal of clinical manifestations [75,77,78]. Hypophosphatemia is considered a cause of reversible CM [55].
Diagnosis and management
Hypophosphatemic CM lacks evidence-based or expert consensus guidelines for diagnosis. Currently, diagnosis is achieved by excluding known causes of CMs such as cardiac ischemia, hypertension, rheumatic heart diseases and congenital abnormalities as well as reversible causes such as alcohol, toxins, infection and metabolic abnormalities [25]. However, current evidence on diagnosis is based on case reports that do not explain confounding factors that may reverse hypophosphatemic CM. Diagnosis for hypophosphatemic CM consists of two steps. The first is to determine the underlying cause through taking detailed patient’s medical history. If the cause is iatrogenic (hypophosphatemia results from the combination of interferon and ribavirin for hepatitis C virus infection, the risk and benefits of the causative treatment must be weighed. Patient history can help determine the cause of hypophosphatemia – decreased intestinal absorption, increased urinary excretion and removal by renal replacement therapies [55].
If patient’s history does not clarify aetiology, urinary phosphate excretion can be measured to assist diagnosis [79]. The 24-hour collection and calculation of fractional excretion of filtered phosphate (FEPO) are valid to determine aetiology. 24-hour urine excretion < 100 mg or FEPO4 < 5% suggests renal phosphate conservation and most likely the patient has internal redistribution or decreased intestinal absorption. A 24-hour urine phosphate excretion > 100 mg or FEPO4 > 5% in the presence of hypophosphatemia suggests renal phosphate wasting indicating hyperparathyroidism or vitamin D deficiency is the cause. Second, once establishing the diagnosis of hypophosphatemia, disease severity should be ascertained. In mild to moderate disease in non-mechanically ventilated patients, oral supplementation is sufficient. In severe hypophosphatemia, intravenous phosphate replacement therapy is often recommended. Phosphate supplementation therapy is associated with reversal of symptoms and cardiac structure and function obviating the need for standard HF medication [53,55].
Overview
Magnesium is the second most abundant intracellular ion. It is naturally present in many foods as well as available as a dietary supplement. Its relationship with CV system, arterial hypertension, central nervous system, skeletal muscles and pregnancy is an established knowledge [80,81]. It is involved in essential metabolic processes including ATP-dependent biochemical reactions, synthesis of DNA, RNA expression, cell signalling at muscle and nerve levels, and glucose and blood pressure control, among others [82]. In the human body, high magnesium concentration occur in bones, and only 1% in the serum and 31% in the intracellular space diluted in the cytoplasm or conjuncted to enzymes or ATP [80]. The usual daily magnesium consumption is 200 to 300 mg but although only a third of this quantity is absorbed through the small intestine. Renal excretion rate of magnesium is 100 mg/day while normal serum values for magnesium is 0.75 to 1.5 mmol/L and values less than 0.75 mmol/L is considered hypomagnesium [80]. Magnesium plays an essential role in mitochondrial integrity, oxidative phosphorylation, protein synthesis, nucleic acid stability, membrane permeability and neuro-muscular excitability [81]. Hypomagnesium has been shown to induce other electrolyte imbalance conditions including hypocalcaemia, hypokalaemia and hypophosphatemia. Since magnesium is not routinely measured, its significance in CV disease (CVD) is often overlooked. A number of studies have described hypomagnesium and CVD including atherosclerosis, ischemic heart disease, and coronary risk factors such as coronary artery spasms, myocardial infarction, thrombosis and arrhythmias. However, hypomagnesium as a cause of CM is uncommon and very rare [83,84].
Evidence based on animal studies
Hypomagnesium has been implicated as a cause of CM in both human and animal studies [81,85-88]. In animal models, hamsters fed with a magnesium deficient diet developed CM with foci of myocardial necrosis, calcification and modest mononuclear and giant cell infiltration. Moreover, hamsters administered with nifedipine had a hose-dependent reduction in lesion abundance and diameter whereas hamster administered with digoxin produced a dose-dependent increase in lesion abundance and diameter [84]. These findings support the hypothesis that lesions are secondary to calcium overload following an increase in myocardial sodium due to the inhibition of the Na+-K+-ATPase and secondary sodium, calcium exchange in magnesium deficient state [88]. In a related study involving Syrian male hamsters fed on a magnesium-deficient diet or identical diet supplemented with MgCl were found more susceptible to ischaemia-induced cardiac injury in the setting of magnesium deficient [86]. Production and effects of catecholamines intensify during cellular magnesium depletion. The detrimental effect of high catecholamines levels and magnesium deficiency maybe synergistic in the myocardium [83]. In rabbits, magnesium supplementation reduces ultrastructural features of myocardial injury caused by epinephrine injection without an effect on changes in intracellular distribution of calcium-induced by epinephrine [89].
Evidence based on human studies
Human studies have also supported the role of magnesium deficiency in the development of CM. Patients with hypoparathyroidism can manifest CM, which responds to a dual supplementation therapy of magnesium and calcium [90]. Magnesium deficiency and CM are also commonly observed in patients with chronic or heavy alcohol consumption [91]. Individuals living in low magnesium equatorial regions and those consuming magnesium-deficient diet have been shown to develop spontaneous endomyocardial fibrosis of undetermined aetiology [91-93]. Post-mortem examination of their hearts reveal low concentrations of magnesium and high concentrations of thorium and cerium, which are thought to be cardiotoxic [83]. Kurnik et al. [81] reported a patient with bulimia who presented with magnesium deficiency resulting in refractory and eventually fatal CM. Despite evidence associating hypomagnesium with CM, there is need for clinical trials to clarify the pathogenic role of magnesium deficiency with CM in humans.
Overview
The association between renal abnormalities (or chronic kidney disease [CKD]) and CVD is well-recognized and well-documented [94-98]. The term CKD encompasses all renal diseases (abnormalities) from the earliest stages through to end-stage renal disease (ESRD) that requires renal replacement therapy [98]. Uraemia (uremic syndrome) is a serious complication of CKD and acute kidney injury characterized by accumulation of urea and other waste products in the body due to the inability of the kidney to eliminate them [98]. In uraemic patients treated by dialysis, CVD accounts for >50% of mortality and the incidence of deaths is 5-10 fold greater in uraemic patients compared to age-matched general population [96,99]. The major cause of CVD-associated death is myocardial ischemia although death may also due to HF and sudden cardiac death (SCD) [99]. Although most of the early evidence on CVD in CKD focussed on ESRD, recent evidence suggests CVD risk increases very early in the natural history of CKD. The risk of CVD death in early stage CKD far exceeds the risk of progressing to dialysis [94]. In CKD patients, typical echocardiography-defined cardiac abnormalities include systolic dysfunction, LV dilatation and LV hypertrophy (LVH) [100]. These complications are a consequence of the underlying CKD-related CM, which has been termed uraemic CM [100-102]. Although echocardiography reveals several CVD abnormalities (LVH, LV dilatation, LV systolic and diastolic dysfunction) in uraemic CM patients [103], Cardiac magnetic resonance imaging (CMRI) suggests LVH is the primary manifestation [104,105]. Thus, uraemic CM may be described as pathological cardiac hypertrophy in the setting of impaired renal function [96].
Pathophysiology
The pathophysiology of uraemic CM CKD or ERSD patients is multifactorial, but primarily involving LVH as well as other LV abnormalities [95,96]. Among ESRD patients, about 73.4% of those started on dialysis have LVH, 35.8% have LV dilatation and 14.8% have LV systolic dysfunction [100]. Usually, LVH does not regress or even aggravates with time on dialysis and its presence is associated with high risk or mortality and CV events including SCD [99]. LVH is the primary pathologic feature of uraemic CM associated with a reduction in capillary density, creating an imbalance between oxygen demand and supply leading to ischemia. In turn, ischemia promotes cardiomyocyte apoptosis as well as extracellular matrix and collagen accumulation leading to interstitial fibrosis, which induces LV stiffness, increased LV filling pressure, impaired diastolic filling and diastolic dysfunction. Furthermore, myocardial fibrosis aggravates ischemia by reducing capillary density and coronary reserve and increases the risk of ventricular arrhythmias and SCD. Comorbid coronary disease, common in CKD and ESRD patients further contributes to ischemia, myocardial damage and fibrosis [95-100].
In CKD and ERSD patients, LVH manifests as a consequence of pressure overload, volume overload and the uraemic state itself [96]. LV pressure overload frequently results from hypertension and arteriosclerosis and occasionally from aortic stenosis. LV volume overload results from arteriovenous (AV) fistula, anaemia and hypervolemia. LV pressure or volume overload leads to the development of LVH. From a hemodynamic view, LVH is an adaptive remodelling process of the LV, which compensates the increased cardiac work induced by increased afterload, increased preload or both. However, continued LV overload may lead to maladaptive changes and death to the cardiomyocytes exacerbated by decreased perfusion, malnutrition, uraemia and hyperparathyroidism [96-98]. Cardiomyocyte loss may result in LV dilatation and ultimately systolic dysfunction. In addition, myocardial fibrosis may occur causing electrical derangement, which further diminishes cardiac compliance leading to diastolic dysfunction. Thus, while LVH is the primary manifestation of uraemic CM, more advanced disease can present with LV dilatation accompanied with systolic dysfunction and depressed LVEF [96]. Animal and human studies implicate uraemic state itself as the cause of the development of LVH, regardless of the pressure and volume overload. Correction of hypertension in rats with renal injury does not prevent LVH while subjecting renal injury to rats LVH develops [106,107]. In humans, LVH develops in high-risk populations with kidney disease despite effective control of hypertension and non-diabetic patients with known diabetic nephropathy developed blood-pressure independent LVH [108,109].
Recent evidence suggest that non-hemodynamic factors, principally hyperphosphatemia, may play a role in the pathophysiology of uraemic CM via inducing vascular calcification. Indeed, phosphotoxicity is considered the main cause of CVD mortality [110]. Vascular calcification is thought to be an active process regulated by cells, where ectopic deposition of calcium and phosphate salts occurs particularly in the muscular layers of arteries and heart valves [111]. Hyperphosphatemia induces vascular calcification via acting directly on type III Na+ dependent phosphate co-transporter of vascular smooth muscle cells thereby increasing intracellular phosphate level and activating bone formation-related gene expression [111]. Hyperphosphatemia is also toxic to endothelial cells (ECs) and may cause endothelial dysfunction, release of endothelial membrane micro-particles and induction of ECs apoptosis [112]. Although there is no direct evidence linking hyperphosphatemia with LVH, several studies demonstrate cardiomyocyte hypertrophy and myocardial fibrosis in animal fed with high-phosphate diet [102]. Hyperphosphatemia may lead indirectly to LVH by inducing hypertension, increased pulse pressure, cardiac afterload and apoptosis. Recently, high FGF-23 levels and αKlotho deficiency have been suggested to play a role in the pathophysiology uraemic CM but their precise roles have not been clarified [95].
Diagnosis
Diagnosis of uraemic CM lacks specific guidelines although it may borrow from diagnostic methods used on CKD and ESRD patients with HF, which consists of cardiac imaging (by echocardiography and/or CMRI) and the assessment of serum levels of natriuretic peptides [113]. For CKD patients presenting with cardiac symptoms, 2D echocardiography including Doppler imaging is useful for assessing LV structure and function and for the diagnosis of the cause of LV dysfunction such as LVH, myocardial ischemia, valvular disease and pericardial effusion or constriction [114]. Echocardiography provides measurements of ventricular diameters and volumes, wall thickness, chamber geometry, LVEF and regional wall motion abnormalities as well as in the diagnosis of systolic (depressed LVEF) versus diastolic dysfunction (HF with preserved LVEF) [115]. For ERSD patients, the kidney disease outcomes quality initiative (KDOQI) guidelines recommend echocardiograms to be performed 1-3 months after initiation of dialysis and every 3 years thereafter regardless of symptoms [116]. CKD patients with significantly depressed LV systolic function should undergo evaluation for coronary artery disease using stress echocardiography, nuclear imaging or computed tomography angiography, or coronary angiography as recommended by KDOQI [116]. Cardiac MRI enables accurate evaluation of LV and LA volume and can detect myocardial viability and scar tissue [117]. However, cost and availability, as well as risk of nephrogenic systemic fibrosis from gadolinium contrast in CKD patients, it is not recommended as a routine imaging tests in CKD patients [114].
Laboratory tests for serum levels of natriuretic peptides (NPs) may provide diagnostic clue to prompt further imaging tests among HF patients, although there is no reliable evidence for their diagnostic and prognostic value or in guiding treatment in CKD or ERSD patients [114]. Ideally, atrial natriuretic peptides (ANP) and B-type natriuretic peptide (BNP) produced by atrial and ventricular myocytes is in response to an increase in atrial or ventricular diastolic filling pressure and wall distention [118,119]. Serum levels of BNP and NT-proBNP reflect LV wall stress [118] and are greatly increased in HF patients and strongly correlate with severity of systolic and diastolic dysfunction as well as severity of HF as assessed by the New York Heart Association functional class [118,120-122]. BNP and NT-proBNP are also important independent prognostic markers of mortality and other cardiac endpoints in HF patients [60]. In CKD and ERSD, impaired renal clearance affect serum levels of NPs but still maintain a string relationship with LV end-diastolic wall stress [123]. In patients on dialysis, serums NPs show significant associations with LVH, LV systolic and diastolic dysfunction, and LA dilatation [123-127]. ECG abnormalities such as prolonged QT interval, increased QT dispersal, frequency of ectopic and tachyarrhythmias are frequently observed in CKD and ERSD patients [114].
Treatment
In uraemic CM, the predominant pathological feature is LVH, which is an independent predictor of survival and regression of LV is associated with reduced CV risk and improved survival [128-130]. Conventional haemodialysis is the most common treatment for uraemic CM associated with reduction in LVH [131]. Dialysis, a mainstay treatment for patients with renal failure, has been described to reverse systolic dysfunction and improve LVEF in some ESRD patients [113]. Supplementary medication can include the contemporary HF medication. ACE-I has been shown to be effective even in normotensive patients. Ramipril for instance reduces LV mass in dialysis patients who were normotensive prior to therapy [132]. However, the beneficial effect of ACE-I beyond lowering blood pressure remains unclear although neuroendocrine mechanisms are thought to be implicated [133,134].
Meta-analysis of diagnosis of uraemic CM
Diagnosis and treatment of electrolyte imbalance and uraemic CMs lack specific guidelines. In particular, there are no clinical trials on electrolyte imbalance CM and the current evidence comes from case reports whose outcomes are less reliable relative to clinical trials as well as cannot form the basis of a meta-analysis. In the few trials mentioning electrolyte imbalance, it is discussed within the broader context of DCM, which does not provide specific knowledge on diagnosis and treatment. Thus, further prospective large-scale clinical trials are warranted to clarify the current diagnostic and therapeutic strategies of electrolyte imbalance CM. Similarly, diagnosis and treatment of uraemic CM remains a clinical challenge due to heterogeneity of clinical features often leading to delayed diagnosis. LVH based on ECG or echocardiography is a typical diagnostic feature of uraemic CM although in patients with subclinical disease (have preserved systolic function and do not exhibit cardiac symptoms) diagnosis is often delayed. Recently, speckle tracking echocardiography (STE) and CMRI have shown promise to allow for early diagnosis as well as improve the accuracy of diagnosis. The STE modality is able to detect subclinical uraemic CM while CMRI to improve diagnosis and prognosis by detecting LVH and LV dilatation, which are often over-estimated on echocardiography. Thus, this section performs a meta-analysis of diagnostic methods for uraemic CM based on published data from clinical trials on CKD and ERSD patients.
The search for relevant studies was conducted on PubMed, Cochrane and Google Scholar. Key search terms used were uraemic cardiomyopathy and all its variants (chronic kidney disease, end stage renal disease, heart failure) and diagnosis (echocardiography, cardiac magnetic resonance, electrocardiography, serum natriuretic peptides). Additional manual search was conducted on reference lists of the included studies. There was no language restriction. Search results were screened by title, abstract and then full text to identify eligible trials fulfilling the inclusion criteria. Studies were considered if they assessed any of the following diagnostic methods – ECG, echocardiography, serum NPs and CMRI, and reported the following outcomes – LV function and structure, serum NPs levels or ECG abnormalities.
Study characteristics
Of 264 studies yielded by the online database search, 18 studies met the inclusion criteria. A further seven (7) studies were identified by manual search of the references of the included studies, bringing the total number of studies forming the final dataset for meta-analysis to 25 [135-160]. The included studies were published between 1999 and 2018. Table 2 provides a summary of the characteristics of the included studies. In total, the 25 studies enrolled a combined patient population of 2,861, with mean age 50.42 (range 13.7 to 61.9 years) and a male preponderance (61.9%). Criteria for patient inclusion in the individual studies were patients with renal abnormalities - diagnosed with CKD or ERSD, pre-dialysis (evaluated for haemodialysis) or on haemodialysis thrice weekly and/or on renal transplant recipients. In particular, STE studies included patients with preserved LV systolic function (LVEF) and without signs and symptoms of cardiac dysfunction. Nine (9) clinical trials evaluating the diagnostic role of STE included control population totalling 337 healthy (non-CKD or ERSD) individuals for comparison with uraemic patients [145,149,150,152,154,156-159]. A greater majority of the studies (n=16; 64%) evaluated either echocardiography (conventional, Doppler or STE) alone or in combination with ECG or CMRI [141,142,145,148-160], and six (6) each (48%), provided diagnostic data on CMRI [141-146] and ECG [136-141].
Table 2. Summary of characteristics of the included studies
Author [Ref #] |
Year |
Patient No. |
Male (n) |
Mean Age (yrs.) |
Criteria for patient selection |
Diagnostic Test |
Summary of Key Findings |
Hage [136] |
2010 |
280 |
173 |
53±9 |
ESRD patients evaluated for kidney Tx |
ECG |
QTc occurs in 39% of ESRD patients and an independent prognosticator of mortality, with a 5-year death rate at 47% and survival (HR: 1.008; 95% CI: 1.001-1.014, p=0.016) |
De Bie [137] |
2012 |
277 |
172 |
56.3±17 |
Patients on chronic HD > 3 months |
ECG |
Abnormal QRS-T angle calculated from 12-ECG (≥1300 in men and ≥1160in women) was associated with higher risk of all-cause death (HR: 2.99; 95% CI: 1.04-8.60) |
Genovesi [138] |
2013 |
122 |
79 |
71.3 |
Patients on HD |
ECG |
Prolonged QTc (>450 ms in men; >460 ms in women) occur in 36% of patients, and an independent predictor of all-cause mortality (HR: 2.16: 95% CI: 1.20-3.91) and SCD (HR: 8.33; 95% CI: 1.71-40.48) |
Tereshchenko [139] |
2016 |
358 |
211 |
55±13 |
ESRD patients on HD |
ECG |
QTc ≥750 was associated with increased CV mortality (HR: 2.99; 95% CI: 1.31-6.82) and SCD (HR: 4.52; 95% CI: 1.17-17.40) |
Shafi [140] |
2017 |
124 |
72 |
49.9±13.8 |
CKD patients not on renal replacement therapy |
ECG |
ECG abnormality occurred in 78.4% of all CKD patients: LVH (40%), Q waves (27.2%), ST-segment changes (17.6%), prolonged QRS duration (19.2%), and tachycardia (17.6%) |
Stewart [141] |
2005 |
55 |
33 |
46.8±14.1 |
Non-diabetic renal disease on HD |
ECG, Echo |
Incidence of LVH correlated with renal function, ranging from 39% in near normal renal function to 80% with renal allograft. LVMI is also highest in HD patients |
Stewart [142] |
1999 |
32 |
21 |
NA |
Patients undergoing HD 3 times per week |
CMRI, Echo |
LV hypertrophy and dilatation are common in HD patients. Echo significantly overestimates LV mass relative to CMRI in the presence of LVH and LV dilatation |
Mark [143] |
2006 |
134 |
92 |
52.2±10.4 |
ESRD patients evaluated for renal Tx |
CMRI |
14.2% patients had sub-endocardial LGE, 14.2% diffuse LGE, and 28.4% myocardial fibrosis. LVH is predominant to uraemic CM while LVH and dilation are due to underlying ischemic heart disease. |
Patel [144] |
2009 |
246 |
157 |
51.4±12.1 |
Patients on HD 3 times weekly for > 3 months |
CMRI |
LVH occur in 63.8% of patients. Both LVH and LVM are associated with higher end diastolic and systolic volumes |
Ebeid [145] |
2017 |
30 |
15 |
41.3±11.6 |
ESRD patients on maintenance HD |
CMRI, Echo |
Echo overestimated LVM and LVMI than CMRI as well as LVH detection (66.6%) compared to 36.7% in CMRI. |
Rutherford [146] |
2017 |
24 |
15 |
61.2±13.4 |
Patients undergoing HD for < 12 months |
CMRI |
CMRI reveals after 6 months HD a reduction in LVMI (78.8 to 69.9 g/m2), improved GLS (-17.9 to -21.6%) and troponin T (38.8 to 30.8 pg/L) |
Gong [147] |
2018 |
40 |
28 |
56±11 |
Patients with ESRD and kidney Tx |
CMRI |
In 12 months, kidney Tx is associated with improvement in GCS and GRS correlate with reduction in LVEDV index and LVESV index |
Pecoits-Filho [148] |
2010 |
50 |
NR |
62.8±2.2 |
Uraemic CM patients undergoing HD |
2D-Echo |
Uremic CM patients have higher mean natriuretic peptides levels (NT-proBNP, BNP, ANP). Post HD NT-proBNP increased by 14%, BNP and ANP decreased by 17% and 56%. |
Altekin [149] |
2014 |
87 |
NR |
NR |
ESRD patients with preserved ejection fraction on HD |
2D-Echo STE |
Compared to controls, ESRD patients have significantly reduced GRS (45.17±17.28 vs. 53.97±14.29); GLS (-19.71±3.1 vs. -30.13±2.1) but preserved LVEF (64.39±5.7%) due to preserved GCS (19.42±7.14 vs. 18.57±4.12, p=0.155) |
Chen [150] |
2014 |
35 |
19 |
52.7±7.6 |
CKDP patients on HD and uraemia |
2D-Echo STE |
3D-STE may detect myocardial dysfunction (SD strain and regional longitudinal strain) in uraemia patients on HD with preserved LVEF (57.8±1.9%) |
Kramann [151] |
2014 |
171 |
111 |
NR |
ESRD patients on dialysis eligible for STE |
2D-Echo STE |
Over 2.1±0.9 year follow-up 44% died. Predictors of mortality: LVEF (HR: 0.97; 95% CI: 0.95-0.99); GLS (HR: 1.17; 95% CI: 1.07-1.28) |
Panoulas [152] |
2014 |
39 |
21 |
57.4±14.6 |
CKD patients not on HD and with preserved EF |
2D-Echo STE |
In CKD without CV symptoms and preserved LVEF, STE can identify subclinical abnormalities decreased GLS (-18.01±2.64 vs. -20.67±3.06 control) and LV twist (24.6±5.35 vs. 16.2±4.8 control) |
Krishnasamy [153] |
2015 |
183 |
105 |
55±15 |
Patients with ESRD and/or on HD for > 3 months |
2D-Echo STE |
Over 7.8 years follow-up, 61.2% died. GLS is a predictor of all-cause mortality (HR: 1.09; 95% CI: 1.02-1.16) and CV mortality (HR: 1.16; 95% CI: 1.04-1.30). Impaired GLS > -16% has 5.6 fold increased in the risk for CV mortality in patients with preserved LVEF |
Ali [154] |
2016 |
49 |
32 |
42.9±15.13 |
Uraemic patients with preserved systolic function on HD |
2D-Echo STE |
RV and LV longitudinal strains are significantly lower in patients than in controls (-9.6 vs. -15.3 for RV and -11.3 vs.-14.8 for LV) despite normal systolic function. |
Ibrahim [155] |
2016 |
26 |
12 |
45.4±16.6 |
CKD patients on dialysis |
2D-Echo STE |
After HD there is significant reduction in peak systolic GLS in ling axis (-19.32±4.3 to -16.58±3.87), GLS in apical 4-chamber view (-17.73±4.76 to -15.98±3.7) and GLS average (-18.59±3.96 to -16.45±3.31) |
Hassanin [156] |
2016 |
90 |
NA |
49.3±14 |
Patients in various stages of CKD |
2D-Echo STE |
Compared to controls. CKD patients have reduced LV longitudinal strain (-16.9±3.8 vs. -22.5±0.6), early diastolic strain rate (1.6±0.5% vs. 2.3±0.2%) and late diastolic strain rate (1.3±0.4% vs. 1.9±0.1%) despite preserved LVEF |
Van Huis [157] |
2016 |
36 |
33 |
15.1 (1.2-17.9) |
ESRD paediatric patients on HD and Tx |
2D-Echo STE |
Interventricular and LV posterior wall thickness are increased in HD and Tx patients and reduced GLS compared to controls with no significant different in LVEF |
Hensen [158] |
2017 |
304 |
200 |
62±14 |
Pre-dialysis and HD CKD patients |
2D-Echo STE |
Over 29 months follow-up, 34% died. LV GLS ≤10.6% showed significantly worse prognosis and increase risk of all-cause mortality (HR: 2.18; 95% CI: 1.17-4.06) |
Ma [159] |
2018 |
31 |
NR |
NR |
Clinically stable uremic outpatients on HD 3 times per week |
2D-Echo STE |
2D-STE cam identify LV myocardial abnormalities (Significantly reduced basal, mod and apical longitudinal strain) in uraemic CM patients with preserved LVEF |
Tamulenaite [160] |
2018 |
38 |
19 |
58.6 (49.8-72.0) |
ESRD patients on chronic HD with preserved LVEF on echo |
2D-Echo STE |
ESRD patients on HD had preserved LVEF and lower LV GLS (-22.43±2.71 vs. -24.73±2.03); GCS at mitral valve (-18.73±3.49 vs. -21.67±2.22) and GCS at papillary muscles (-18.64±2.75 vs. -25.45±2.48 |
CKD: Chronic Kidney Disease; CM: Cardiomyopathy; CMRI: Cardiac Magnetic Resonance Imaging; CV: Cardiovascular; ECG: Electrocardiography; ESRD: End Stage Renal Disease; GLS: Global Longitudinal Strain; GCS: Global Circumferential Strain; GRS: Global Radial Strain; HD: Haemodialysis; LVEF: Left Ventricular Ejection Fraction; NR: Not Reported; STE: Speckle Tracking Echocardiography; Tx: Transplantation
Study outcomes
In a pooled analysis of three studies [136,137,140], LVH based on ECG’s Cornell voltage criteria was the most common diagnostic feature (ECG abnormality) that occurred in 237 out of 681 patients (Event rate [ER]: 34.8%; 95% CI: 19.0 to 54.8). Prolonged QTc (> 450 ms) pooled from three studies [136,138,140] was also common in 162 out of 526 patients (ER: 25.0%; 95% CI: 12.1 to 44.6). Other less common ECG abnormalities reported in two studies (136,140) is prolonged QRS duration (> 100 ms) in 75 out of 404 patients (ER: 18.6%; 95% CI: 15.1 to 22.7) and prolonged PR interval (> 200 msec) in 43 out of 404 patients (ER: 14.7%; 95% CI: 11.1 to 19.2) (Table 3). In four studies [136,137,140,141], the weighted mean of QTc is 437 ms (Figures 1 and 2) and QRS duration is 97 ms
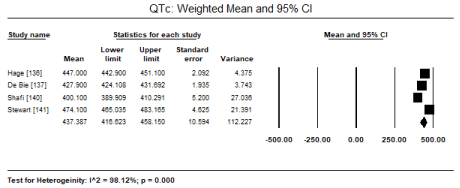
Figure 1. Weighted mean and 95% CI for QTc
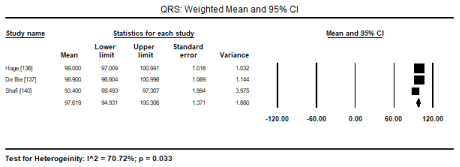
Figure 2. Weighted mean and 95% CI for QRS duration
Table 3. ECG abnormalities in uraemic CM patients
ECG Abnormality |
No. of Studies [Reference #] |
No. of Patients |
Event Rate (%) |
95% CI |
LVH |
3 [136,137,140] |
237/681 |
34.8 |
19.0 to 54.8 |
Prolonged QTc |
3 [136,138,140] |
165/526 |
25.0 |
12.1 to 44.6 |
Prolonged QRSd |
2 [136,140] |
75/404 |
18.6 |
15.1 to 22.7 |
Prolonged PR Interval |
2 [136,140] |
43/404 |
14.7 |
11.1 to 19.2 |
LVH: Left Ventricular Hypertrophy; QRSd: QRS Duration
Pooled data from 11 echocardiographic studies [145,148-150,153-155,157-160] reveal they enrolled CKD and ERSD patients on haemodialysis with preserved systolic function (weighted mean LVEF: 57.65%; 95% CI: 55.16 to 60.14). In four studies [152,163,155,157] there were fewer cases of LV hypertrophy found in 107 out of 284 patients (ER: 26.2%; 95% CI: 13.6 to 44.5). In two CMRI studies [143,144], pooled data reveals higher cases of LVH (ER: 67.3%; 95% CI: 59.2 to 74.4). Compared to healthy controls, there is a tendency towards increased LVEDD on echocardiography in uraemic CM patients (WMD: 4.536 mm; 95% CI: -0.865 to 9.937; p = 0.100: Figure 3) [150,154,157,159,160]. On Doppler echocardiography, there was a trend towards decreased E/A ratio (a non-significant decrease) among uraemic patients compared to controls (WMD: -0.145; 95% CI: -0.371 to 0.081; p = 0.209; Figure 4) [150,154,157,159,160] and significantly increased E/E; ratio (WMD: 3.227; 95% CI: 2.711 to 3.943; p = 0.000; Figure 5) [150,152,159,160].
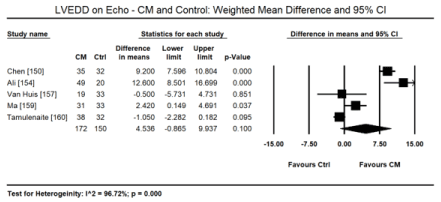
Figure 3. Weighted mean difference LVEDD for CM and control patients
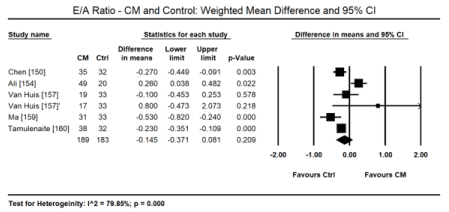
Figure 4. Weighted mean difference EA ratio for CM and control patients
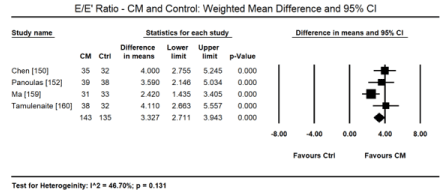
Figure 5. Weighted mean difference EE’ ratio for CM and control patients
In uraemic CM patients with preserved LVEF, STE reveals impairment in the LV longitudinal and circumferential deformation indices compared with age-matched healthy controls. In a pooled data in 8 studies [146,147,150-152,156,157,160], STE detected a small but significant decrease in LV global longitudinal strain (GLS) in uraemic CM patients (WMD: 4.028; 95% CI: 1.921 to 6.136; p = 0.000; Figure 6). In two studies [10,12], GLS was also an independent prognostic indicator of mortality in uraemic CM patients (Hazard ratio [HR]: 1.166; 95% CI: 1.087-1.250; p= 0.000). In a pooled analysis of six studies [147,151,152,156,157,160], there was also a significant decrease in global circumferential strain (GCS) in uraemic CM patients (WMD: 1.234; 95% CI: -0.049 to 1.518; p = 0.060; Figure 7). In four studies [147,149,151,157] there was a tendency towards decreased global radial strain (GRS) in uraemic CM patients (WMD: 2.864; 95% CI: -6.558 to 12.287; p = 0.551; Figure 8).
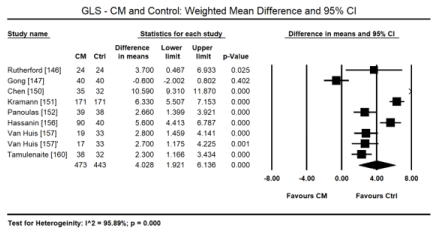
Figure 6. Weighted mean difference GLS for CM and control patients
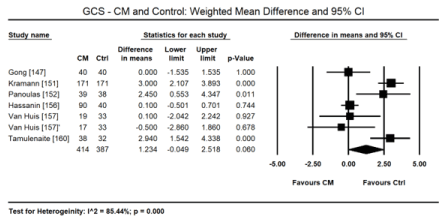
Figure 7. Weighted mean difference GCS for CM and control patients
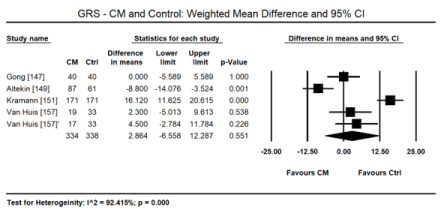
Figure 8. Weighted mean difference GRS for CM and control patients
High mortality and morbidity among CKD patients on dialysis and/or renal transplant remain an important and unresolved clinical issue often manifesting as uraemic CM. in these patients, cardiac abnormalities especially abnormal LV geometry and functions are common and correlate with a high CV mortality and all-cause mortality [161,162]. Early identification of these high-risk patients should allow physicians to optimize therapy, which may lower morbidity and mortality [162]. The present meta-analysis finds ECG, echocardiography and CMRI may play important roles in the diagnosis of uraemic CM. Standard 12-lead ECG (based on Cornell voltage criteria) can detect LVH in 34.8% and prolonged QTc (> 450 ms) in 25% of uraemic CM patients. Prolonged QRS duration (> 100 ms) and PR interval (> 200 msec) are less common, occurring in 18.6% and 14.7% respectively. In asymptomatic uraemic CM patients with preserved LVEF (57.65%), echocardiography reveals fewer cases of LVH (26.2%) while higher on CMRI (67.3%). Echocardiography reveals an increasing trend of LVEDD and decreasing trend on E/A ratio but significantly increased E/E’ ratio in uraemic CM patients. However, STE is merging as echocardiography modality capable of detecting subclinical changes such as impaired LV GLS and GCS in patients with preserved LVEF as well as associates GLS with an increased risk of mortality (HR: 1.166; 95% CI: 1.087-1.250).
The present findings on the value of ECG, echocardiography and CMRI on the diagnosis of uraemic CM patients is consistent with previous review studies. Reviews of echocardiographic studies report a high prevalence of cardiac abnormalities in ESRD patients specifically LVH, systolic and/or diastolic dysfunction and ventricular dilatation, which portend a more ominous prognosis that conventional CV risk factors even when adjusted for coronary disease, which may explain the high incidence of SCD in this population [100,128]. Increased QT interval and dispersal on ECG is associated with LVH, the development of arrhythmias and SCD in ESRD patients including transplant recipients but the association in uraemic CM patients is unknown. In patients with advanced or ESRD, QT interval is increased but QTc has a similar but less marked pattern. In a multivariate analysis, QTc is dependent on LVMI, EF, E/A ratio and LV end diastolic volume [141]. However, studies investigating changes in QTc with haemodialysis reveal mixed results. Over a 30% of dialysis patients have prolonged QTc interval prior to dialysis. Dialysis itself has a heterogeneous effect on QTc. Some indicating prolongation while others no effect possibly because of variation in patient characteristics and co-occurring cardiac comorbidities [163].
Left ventricular EF is the major aim of echocardiographic study used to reflect myocardial contraction strength. It is a longstanding recognized parameter in cardiology used in the evaluation of a wide range of heart conditions. However, patients with subclinical uraemic CM often have preserved LVEF and normal or near normal ventricular dimensions and diameter, which reduces the accuracy of conventional echocardiography in early diagnosis of uraemic CM [164]. The STE modality, a relatively new modality but extensively validated in many cardiac diseases, is emerging as a valuable test for the detection of subclinical cardiac changes. STE works by tracking myocardia speckles displacement, frame-by-frame in an angle dependent way to determine multiple aspects of LV contraction mechanics such as segmental displacement and velocity, strain and strain rate, rotations, twist/torsion and its derivatives [165-167]. A strong correlation between GLS and LVEF (r=0.95; p < 0.001) and GLS rate (r=0.85; p< 0.001) have been described, which is stronger in patients with mild or severe systolic dysfunction compared to those with preserved LV function [164]. Thus, CKD or ERSD patients suspected with cardiac dysfunction should be considered for STE to evaluate for subclinical changes in ventricular structure and function. Recently, the use of 3D-STE is increasing, which promises to address intrinsic limitation of 2D-STE as well as improve reproducibility and accuracy of diagnosis [168]. Despite extensive validation of the STE modality, it remains an evolving technique and improvements such as tracking accuracy are still needed. This accuracy is also highly dependent on image quality since suboptimal resolution can produce negative impact on final results [164].
Besides STE, the use of CMRI can improve the diagnosis of the early stages of CKD, which is often underdiagnosed although the deleterious effect on the CV system are already at work. In Uraemic CM patients, the evaluation of early myocardial damage is essential to prevent major CV events [169,170]. Myocardial fibrosis is one of the major consequences of progressive CKD, which may lead to re-entry arrhythmias and chronic myocardial dysfunction predisposing patients to SCD and/or HF. LGE-CMRI was originally developed to detect chronic infarcted myocardium such as fibrous scar tissue but has also been found useful in the diagnosis of CMs. Subclinical myocardial fibrosis, which has a potential role in the development of uraemic CM, can be measured and characterized by LGE-CMRI modality [169]. The presence of LGE occurs in only 6% of cases of early CKD with no clinical evidence of CVD and 28% in ESRD patients [169]. The new uraemic pattern of LGE may help in differential diagnosis of CKD-related diffuse fibrosis from ischemic scarring. LGE-defined diffuse fibrosis is associated with increased LV mass suggesting that LVH in end-stage CKD is pathological [143]. The main limitation of LGE-CMRI is that it is not sufficiently sensitive to detect diffuse form of myocardial fibrosis since the technique relies relative difference in signal intensities such as considering the lowest myocardial signal intensity as normal regardless of the degree of fibrosis [170]. Finally, severe consequences of gadolinium administration in uraemia (nephrogenic systemic fibrosis) is forcing practitioners to re-evaluate the use of the technique [169].
Electrolyte imbalance and renal abnormalities (uraemia) are less common causes of CMs but potentially reversible following early diagnosis and prompt initiation of appropriate treatment. Despite an ominous diagnosis associated with late diagnosis, electrolyte imbalance and uraemic CMs remain under-appreciated aetiologies of CMs often discussed in the context of dilated CM. Electrolyte imbalance capable of causing CM are hypocalcaemia, hypophosphatemia and hypomagnesaemia. Clinical presentation may vary widely and thus less useful for diagnosis. Diagnosis is by the exclusion of known causes of cardiac dysfunction and demonstration of significantly reduced serum levels of the culprit electrolyte (calcium, phosphate or magnesium). The mainstay treatment of hypocalcaemic, hypophosphatemic and hypomagnesaemic CMs is therapeutic supplementation by calcium, phosphate and magnesium respectively, which may be complemented by conventional HF medication for the management of signs and symptoms of cardiac dysfunction. Uraemic CM results from renal failure often manifesting in CKD or ESRD patients including those on haemodialysis or with renal transplants. Key pathological features are LV hypertrophy, dilatation and systolic dysfunction. Diagnosis for cardiac involvement in CKD or ERSD patients rests on a combination of ECG abnormalities (commonly prolonged QTc), and echocardiography or CMRI to demonstrate LVH, dilatation and systolic dysfunction. STE is promises to detect subclinical LV changes demonstrated by reduced GLS and GCS despite preserved LVEF. Treatment by haemodialysis promises to reverse cardiac function in some patients hut HF treatment remains unclear because current guidelines may not entirely apply to patients with severe renal impairment. Longitudinal or large-scale prospective studies are warranted to clarify diagnosis and treatment of electrolyte imbalance CMs and HF treatment in uraemic CM patients.
- Friedberg CK (1957) Fluid and electrolyte disturbances in heart failure and their treatment. Circulation 16: 437-460. [Crossref]
- Schwinger RH, Erdmann E (1992) Heart failure and electrolyte disturbances. Methods Find Exp Clin Pharmacol 14: 315-325. [Crossref]
- Dei Cas L, Metra M, Leier CV (1995) Electrolyte disturbances in chronic heart failure: Metabolic and clinical aspects. Clin Cardiol 18: 370-376. [Crossref]
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A et al. (2016) Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 37: 1850-1858. [Crossref]
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F et al. (2007) Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29: 270-276.
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D et al. (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 113: 1807-1816.
- Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A et al. (2016) Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134: e579-e646. [Crossref]
- Balci AK, Koksal O, Kose A, Armagan E, Ozdemir F et al. (2013) General characteristics of patients with electrolyte imbalance admitted to emergency department. World J Emerg Med 4: 113-116. [Crossref]
- Lee CT, Guo HR, Chen JB (2000) Hyponatremia in the emergency department. Am J Emerg Med 18: 264-268. [Crossref]
- Shiber JR, Mattu A (2002) Serum phosphate abnormalities in the emergency department. J Emerg Med 23: 395-400. [Crossref]
- Bockenkamp B, Vyas H (2003) Understanding and managing acute fluid and electrolyte disturbances. Current Paediatrics 13: 520-528.
- Ito H, Fujimaki H, Inoue J, Shiraki M (1989) Disorders of fluid and electrolyte metabolism in elderly diabetics. Nihon Ronen Igakkai Zasshi 26: 233-239. [Crossref]
- Goldberg A, Hammerman H, Petcherski S, Zdorovyak A, Yalonetsky S et al. (2004) Prognostic importance of hyponatremia in acute ST-elevation myocardial infarction. Am J Med 117: 242-248. [Crossref]
- Goldberg A, Hammerman H, Petcherski S, Nassar M, Zdorovyak A et al. (2006) Hyponatremia and long-term mortality in survivors of acute ST-elevation myocardial infarction. Arch Intern Med 166: 781-786. [Crossref]
- Fong J, Khan A (2012) Hypocalcemia: updates in diagnosis and management for primary care. Can Fam Physician 58: 158-162. [Crossref]
- Cooper MS, Gittoes NJ (2008) Diagnosis and management of hypocalcemia. BMJ 336: 1298-1302. [Crossref]
- Catalano A, Chilà D, Bellone F, Nicocia G, Martino G et al. (2018) Incidence of hypocalcemia and hypercalcemia in hospitalized patients: Is it changing? J Clin Transl Endocrinol 13: 9-13. [Crossref]
- Katung GK, Akande TS (2016) The prevalence of hypocalcemia in pregnancy and peurperium in northwestern Nigeria. Tropical Journal of Obstetrics and Gynaecology 33: 27-31.
- Holick MF (2007) Vitamin D deficiency. N Engl J Med 357: 266-281. [Crossref]
- Mughal MZ, Salama H, Greenaway T, Laing I, Mawer EB (1999) Lesson of the week: florid rickets associated with prolonged breast feeding without vitamin D supplementation. BMJ 318: 39-40. [Crossref]
- Tolman KG, Jubiz W, Sannella JJ, Madsen JA, Belsey RE et al. (1975) Osteomalacia associated with anticonvulsant drug therapy in mentally retarded children. Pediatrics 56: 45-50. [Crossref]
- Hahn TJ, Hendin BA, Scharp CR, Haddad JG Jr (1972) Effect of chronic anticonvulsant therapy on serum 25-hydroxycalciferol levels in adults. N Engl J Med 287: 900-904. [Crossref]
- Marx SJ (2000) Hyperparathyroid and hypoparathyroid disorders. N Engl J Med 343: 1863-1875. [Crossref]
- Bastepe M, Juppner H (2003) Pseudo-hypoparathyroidism and mechanisms of resistance toward multiple hormones: molecular evidence to clinical presentation. J Clin Endocrinol Metab 88: 4055-4058. [Crossref]
- Peter R, Mishra V, Fraser WD (2004) Severe hypocalcaemia after being given intravenous bisphosphonate. BMJ 328: 335-336. [Crossref]
- Rosen CJ, Brown S (2003) Severe hypocalcemia after intravenous bisphosphonate therapy in occult vitamin D deficiency. N Engl J Med 348: 1503-1504. [Crossref]
- Aguiar P, Cruz D, Ferro RR, Peixoto L, Araujo F et al. (2013) Hypocalcemic cardiomyopathy. Rev Port Cardiol 32: 331-335. [Crossref]
- Kadeli D, Keshava R, Aniyathodiyil G, Somashekara RKS, Magesh B et al. (2018) Cardiomyopathy secondary to hypocalcemia. Journal of the Association of Physicians of India 66: 99-100.
- Cheng Z, Yadav SK, Liu X, Yi Q (2019) A reversible hypocalcemic dilated cardiomyopathy caused by primary hypoparathyroidism. Asian J Med Sci 10: 65-68.
- Rangel I, Barbosa G, de Sousa C, Almeida PB, Campelo M et al. (2012) Hypocalcemia as a reversible cause of heart failure. Rev Port Cardiol 3: 39-41. [Crossref]
- Bansal B, Bansal M, Bajpai P, Garewal HK (2014) Hypocalcemic cardiomyopathy-different mechanisms in adult and pediatric cases. J Clin Endocrinol Metab 99: 2627-2632. [Crossref]
- Szent-Gyorgyi AG (1975). Calcium regulation of muscle contraction. Biophys J 15: 707-723. [Crossref]
- Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU (2008) Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology 149: 558-564. [Crossref]
- Gezmish O, Tare M, Parkington HC, Morley R, Porrello ER et al. (2010) Maternal vitamin D deficiency leads to cardiac hypertrophy in rat offspring. Reprod Sci 17: 168-76. [Crossref]
- Fall T, Shiue I, Bergeå af Geijerstam P, Sundström J et al. (2012) Relations of circulating vitamin D concentrations with left ventricular geometry and function. Eur J Heart Fail 14: 985-991. [Crossref]
- Pilz S, Marz W, Wellnitz B, Seelhorst U, Fahrleitner-Pammer A et al. (2008) Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab 93: 3927-3935. [Crossref]
- Rampe D, Lacerda AE, Dage RC, Brown AM (1991) Parathyroid hormone: an endogenous modulator of cardiac calcium channels. Am J Physiol 261: H1945-H1950. [Crossref]
- Jung YJ, Kim SE, Hong JY, Lee JH, Park DG et al. (2013) Reversible dilated cardiomyopathy caused by idiopathic hypoparathyroidism. Korean J Intern Med 28: 605-608. [Crossref]
- Lee SW, Jung YK, Jeon U, Park SH, Lee SJ et al. (2013) Takotsubo cardiomyopathy associated with severe hypocalcemia secondary to idiopathic hypoparathyroidism. Korean Circ J 43: 573-577. [Crossref]
- Tomar M, Radhakrishnan S, Shrivastava S (2010) Myocardial dysfunction due to hypocalcemia. Indian Pediatr 47: 781-783. [Crossref]
- Price DI, Stanford LC Jr, Braden DS, Ebeid MR, Smith JC (2003) Hypocalcemic rickets: an unusual cause of dilated cardiomyopathy. Pediatr Cardiol 24: 510-512. [Crossref]
- Gupta P, Tomar M, Radhakrishnan S, Shrivastava S (2011) Hypocalcemic cardiomyopathy presenting as cardiogenic shock. Ann Pediatr Cardiol 4: 152-155. [Crossref]
- Kumar M, Saikia D, Kumar V, Tomar R (2011) Vitamin D deficiency presenting with cardiogenic shock in an infant. Ann Pediatr Cardiol 4: 207-209. [Crossref]
- Maiya S, Sullivan I, Allgrove J, Yates R, Malone M et al. (2008) Hypocalcaemia and vitamin D deficiency: an important, but preventable, cause of life-threatening infant heart failure. Heart 94: 581-584. [Crossref]
- Soliman A, Salama H, Alomar S, Shatla E, Ellithy K et al. (2013) Clinical, biochemical, and radiological manifestations of vitamin D deficiency in newborns presented with hypocalcemia. Indian J Endocrinol Metab 17: 697-703. [Crossref]
- Elidrissy AT, Munawarah M, Alharbi KM (2013) Hypocalcemic rachitic cardiomyopathy in infants. J Saudi Heart Assoc 25: 25-33. [Crossref]
- Marwaha RK, Tandon N, Chopra S, et al. (2011) Vitamin D status in pregnant Indian women across trimesters and different seasons and its correlation with neonatal serum 25-hydroxyvitamin D levels. Br Nutr 106: 1383-1389. [Crossref]
- Pourdjabbar A, Dwivedi G, Haddad H (2013) The role of vitamin D in chronic heart failure. Curr Opin Cardiol 28: 216-222. [Crossref]
- Amin A, Minaee S, Chitsazan M, Naderi N, Taghavi S, Ardeshiri M (2013) Can vitamin D supplementation improve the severity of congestive heart failure? Congest Heart Fail 19: E22-E28. [Crossref]
- Malabanan A, Veronikis IE, Holick MF (1998) Redefining vitamin D insufficiency. Lancet 351: 805-806. [Crossref]
- Shoback D (2008) Clinical practice: hypoparathyroidism. N Engl J Med 359: 391-403. [Crossref]
- Bilezikian JP, Khan A, Potts Jr JT, Brandi ML, Clarke BL et al. (2011) Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment and challenges for future research. J Bone Min Res 26: 2317-2337. [Crossref]
- Moreland KJ (1989) Hypophosphatemia: Causes and clinical consequences. J Vet Intern Med 3: 149-159. [Crossref]
- Amanzadeh J, Reilly RF Jr (2006) Hypophosphatemia: an evidence-based approach to its clinical consequences and management. Nat Clin Pract Nephrol 2: 136-148. [Crossref]
- Ariyoshi N, Nogi M, Ando A, Watanabe H, Umekawa S (2016) Hypophosphatemia-induced cardiomyopathy. Am J Med Sci 352: 317-323. [Crossref]
- Knochel JP, Jacobson HR (1986) Renal handling of phosphorus, clinical hypophosphatemia, and phosphorus deficiency. In Brenner BM, Rector FC. The kidney, 3rd ed. Philadelphia: WB Saunders Company 619-622.
- Lotz M, Zisman E, Bartter FC (1968) Evidence for a phosphorous-depletion syndrome in man. N ENgl J Med 278: 409-415. [Crossref]
- Shields HM (1978) Rapid fall of serum phosphorous secondary to antacid therapy. Gastroenterology 75: 1137-1141. [Crossref]
- Juan D, Elrazak MA (1979) Hypophosphatemia in hospitalized patients. JAMA 242: 163-164. [Crossref]
- Agus ZA, Goldfarb S (1982) Calcium and phosphate disorders. Med Clin North Am 65: 393-396. [Crossref]
- Knochel JP (1985) Complications of total parenteral nutrition. Kidney Int 27: 489-496. [Crossref]
- Mostellar ME, Tuttle EP (1964) Effects of alkalosis on plasma concentration and urinary excretion of inorganic phosphate in man. J Clin Invest 43: 138–149. [Crossref]
- Hoppe AN, Metler MA, Berndt TJ, Knox FG, Angielski ST (1982) Effect of respiratory alkalosis on renal phosphate excretion. Am J Physiol 243: F471-F475. [Crossref]
- Murer H, Forster I, Biber J (2004) The sodium phosphate cotransporter family SLC34. Pflugers Arch 447: 763-767. [Crossref]
- Kadkhodayan A, Coggan AR, Peterson LR (2013) A “PET” area of interest: myocardial metabolism in human systolic heart failure. Heart Fail Rev 18: 567-574. [Crossref]
- Lichtman MA, Miller DR, Cohen J, Waterhouse C (1971) Reduced red cell glycolysis, 2,3-diphosphoglycerate and adenosine triphosphate concentration, and increased hemoglobin-oxygen affinity caused by hypophosphatemia. Ann Intern Med 74: 562-568. [Crossref]
- Bersin RM, Kwasman M, Lau D, Klinski C, Tanaka K et al. (1993) Importance of oxygenhaemoglobin binding to oxygen transport in congestive heart failure. Br Heart J 70: 443-447. [Crossref]
- Shibata K, Fujita S, Morita H, et al. (2013) Association between circulating fibroblast growth factor 23, alpha-Klotho, and the left ventricular ejection fraction and left ventricular mass in cardiology inpatients. PLoS One 8: e73184. [Crossref]
- Hu CY, Lee BJ, Cheng HF, et al. (2015) Acetazolamide-related lifethreatening hypophosphatemia in a glaucoma patient. J Glaucoma 24: e31-33. [Crossref]
- Osuka A, Matsuoka T, Idoguchi K. (2009) Is this the worst outcome of metabolic syndrome? Hypophosphatemia and resulting cardiac arrest during the treatment of diabetic ketoacidosis with hypertriglyceridemia. Intern Med 48: 1391-1395. [Crossref]
- Lin KK, Lee JJ, Chen HC (2006) Severe refeeding hypophosphatemia in a CAPD patient: a case report. Renal Fail 28: 515-517. [Crossref]
- Cariem AK, Lemmer ER, Adams MG, et al. (1994) Severe hypophosphataemia in anorexia nervosa. Postgrad Med J 70: 825-827. [Crossref]
- Yang HT, Yim H, Cho YS, et al. (2012) Change of serum phosphate level and clinical outcome of hypophosphatemia in massive burn patient. J Trauma Acute Care Surg 73: 1298-1302. [Crossref]
- Davis SV, Olichwier KK, Chakko SC. (1988) Reversible depression of myocardial performance in hypophosphatemia. Am J Med Sci 295: 183-187. [Crossref]
- O’Connor LR, Wheeler WS, Bethune JE. (1977) Effect of hypophosphatemia on myocardial performance in man. N Engl J Med 297: 901-903. [Crossref]
- Ognibene A, Ciniglio R, Greifenstein A, et al. (1994) Ventricular tachycardia in acute myocardial infarction: the role of hypophosphatemia. South Med J 87: 65-69. [Crossref]
- Heames RM, Cope RA. (2006) Hypophosphataemia causing profound cardiac failure after cardiac surgery. Anaesthesia 61: 1211-1213. [Crossref]
- Schwartz A, Brotfain E, Koyfman L, et al. (2014) Association between hypophosphatemia and cardiac arrhythmias in the early stage of sepsis: could phosphorus replacement treatment reduce the incidence of arrhythmias? Electrolyte Blood Press 12: 19-25. [Crossref]
- Bacchetta J, Salusky IB (2012) Evaluation of hypophosphatemia: lessons from patients with genetic disorders. Am J Kidney Dis 59: 152-159. [Crossref]
- Efstratiadis G, Sarigianni M, Gougourelas I (2006) Hypomagnesemia and cardiovascular system. Hippokratia 10: 147-152. [Crossref]
- Kurnik BR, Marshall J, Katz SM (1988) Hypomagnesemia-induced cardiomyopathy. Magnesium 7: 49-53. [Crossref]
- Rosique-Esteban N, Guasch-Ferré M, Hernández-Alonso P, Salas-Salvadó J (2018) Dietary magnesium and cardiovascular disease: A review with emphasis in epidemiological studies. Nutrients 10: 168. [Crossref]
- Purvis JR, Movahed A (1992) Magnesium disorders and cardiovascular diseases. Clin Cardiol 15: 556-568. [Crossref]
- DiNicolantonio JJ, Liu J, O'Keefe JH (2018) Magnesium for the prevention and treatment of cardiovascular disease. Open Heart 5: e000775. [Crossref]
- Bloom S (1988) Magnesium deficiency cardiomyopathy. Am J Cardiov Path 2: 7-17. [Crossref]
- Freedman AM, Atrakchi AH, Cassidy MM, Weglicki WB (1990) Magnesium deficiency-induced cardiomyopathy: protection by vitamin E. Biochem Biophys Res Commun 170: 1102-1106. [Crossref]
- Kurantsin-Mills J, Cassidy MM, Stafford RE, Weglicki WB (1997) Marked alterations in circulating inflammatory cells during cardiomyopathy development in a magnesium-deficient rat model. Br J Nutr 78: 845-855. [Crossref]
- Ahmad A, Bloom S (1989). Sodium pump and calcium channel modulation of Mg-deficiency cardiomyopathy. Am J Cardiovasc Pathol 2: 277-283. [Crossref]
- Herbaczynska-Cedro K, Gajkowska B (1992) Effect of magnesium on myocardial damage induced by epinephrine. Ultrastructural and cytochemical study. Cardioscience 3: 197-203. [Crossref]
- Giles TD, Iteld BJ, Rives KL (1981) The cardiomyopathy of hypoparathyroidism. Chest 79: 225-229. [Crossref]
- Seelig MS (1980) Magnesium deficiency in the pathogenesis of disease: early roots of cardiovascular, skeletal, and renal abnormalities. Springer Science & Business Media.
- Valiathan MS, Kartha CC, Panday VK, et al. (1986) A geochemical basis for endomyocardial fibrosis. Cardiovasc Res 20: 679-682. [Crossref]
- Valiathan SM, Kartha CC (1990) Endomyocardial fibrosis: the possible connexion with myocardial levels of magnesium and cerium. Int J Cardiol 28: 1-5. [Crossref]
- Edwards NC, Moody WE, Chue CD, Ferro CJ, Townend JN et al. (2014) Defining the natural history of uremic cardiomyopathy in chronic kidney disease: the role of cardiovascular magnetic resonance. JACC: Cardiovasc Imaging 7: 703-714. [Crossref]
- Paulo G, Sanders-Pinheiro H, de Paula RB (2018) Uremic cardiomyopathy: a new piece in the chronic kidney disease-mineral and bone disorder puzzle. Front Med (Lausanne) 5: 206. [Crossref]
- Alhaj E, Alhaj N, Rahman I, Niazi TO, Berkowitz R et al. (2013) Uremic cardiomyopathy: an underdiagnosed disease. Congest Heart Fail 19: E40-E45. [Crossref]
- Tonelli M, Karumanchi SA, Thadhani R (2016) Epidemiology and mechanisms of uremia-related cardiovascular disease. Circulation 133: 518-536. [Crossref]
- Kunz K, Dimitrov Y, Muller S, Chantrel F, Hannedouche T (1998) Uraemic cardiomyopathy. Nephrol Dial Transplant 13: 39-43. [Crossref]
- Raine AE, Margreiter R, Brunner FP et al. (1992) Report on management of renal failure in Europe. XXII, 1991. Nephrol Dial Transplant 2: 7-35. [Crossref]
- Foley RN, Parfrey PS, Harnett JD et al. (1995) Clinical and echocardiographic disease in patients starting end-stage renal disease therapy. Kidney Int 47: 186-192. [Crossref]
- Fliser D, Schweizer C, Ritz E (1991) How many patients with renal insufficiency survive until end-stage renal failure? Nephrol Dial Transplant 6: 600. [Crossref]
- Harnet JD, Murohy B, Collingwood P, Purchase L, Kent G et al. (1993) The reliability and validity of echocardiographic measurement of LV mass index in haemodialysis patients. Nephron 65: 212-214. [Crossref]
- Jardine AG, McLaughlin K (2001) Cardiovascular complications of renal disease. Heart 86: 459-466. [Crossref]
- Siedlecki A, Muslin AJ (2008) Left ventricular hypertrophy in the setting of chronic kidney disease: mechanisms and treatment. US Nephrol 3: 40-42.
- Mark PB, Johnston N, Groenning BA, et al. (2006) Redefinition of uremic cardiomyopathy by contrast-enhanced cardiac magnetic resonance imaging. Kidney Int 69: 1839-1845. [Crossref]
- Wolf WC, Agata J, Chao L, Chao J (2000) Human tissue kallikrein gene delivery attenuates hypertension, renal injury, and cardiac remodelling in chronic renal failure. Kidney Int 58: 730-739. [Crossref]
- McMahon AC, Dodd SM, Hurst MJ, Raine AE (2002) Prolonged calcium transients and myocardial remodelling in early experimental uremia. Nephrol Dial Transplant 17: 759-764. [Crossref]
- Raev DC (1994) Which left ventricular function is impaired earlier in the evolution of diabetic cardiomyopathy? An echocardiographic study of young type I diabetic patients. Diabetes Care 17: 633-639. [Crossref]
- Nielsen FS, Ali S, Tarnow L, et al. (1998) Beneficial impact of ramipril on left ventricular hypertrophy in normotensive non-albuminuric NIDDM patients. Diabetes Care 21: 804-809. [Crossref]
- Ritter CS, Slatopolsky E (2016) Phosphate toxicity in CKD: the killer among us. Clin J Am Soc Nephrol 11: 1088-100. [Crossref]
- Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, et al. (2000) Phosphate regulation of vascular smooth muscle cell calcification. Circ Res 87: E10-E17. [Crossref]
- Amann K, Tornig J, Kugel B, Gross ML, Tyralla K, El-Shakmak A, et al. (2003) Hyperphosphatemia aggravates cardiac fibrosis and microvascular disease in experimental uremia. Kidney Int 63: 1296-1301. [Crossref]
- Segall L, Nistor I, Covic A (2014) Heart failure in patients with chronic kidney disease: a systematic integrative review. Biomed Res Int 2014: 937398. [Crossref]
- Wang AY, Sanderson JE (2011) Current perspectives on diagnosis of heart failure in long-term dialysis patients. Am J Kidney Dis 57: 308-319. [Crossref]
- Paulus WJ, Tschöpe C, Sanderson JE, Rusconi C, Flachskampf F et al. (2007) How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J 28: 2539-2550. [Crossref]
- K/DOQI Workgroup (2005) K/DOQI clinical practice guidelines for cardiovascular disease in dialysis patients. Am J Kidney Dis 45: S1-153. [Crossref]
- Bello D, Shah DJ, Farah GM, Di Luzio S, Parker M et al. (2003) Gadolinium cardiovascular magnetic resonance predicts reversible myocardial dysfunction and remodeling in patients with heart failure undergoing β-blocker therapy. Circulation 108: 1945-1953. [Crossref]
- Yasue H, Yoshimura M, Sumida H, Kikuta K, Kugiyama K et al. (1994) Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation 90: 195-203. [Crossref]
- Hosoda K, Nakao K, Mukoyama M, Saito Y, Jougasaki M et al. (1991) Expression of brain natriuretic peptide gene in human heart. Production in the ventricle. Hypertension 17: 1152-1155. [Crossref]
- Yamamoto K, Burnett Jr JC, Jougasaki M, Nishimura RA, Bailey KR et al. (1996) Superiority of brain natriuretic peptide as a hormonal marker of ventricular systolic and diastolic dysfunction and ventricular hypertrophy. Hypertension 28: 988-994. [Crossref]
- Maisel AS, Krishnaswamy P, Nowak RM, McCord J, Hollander JE et al. (2002) Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. New Eng J Med 347: 161-167. [Crossref]
- Hunt PJ, Richards AM, Nicholls MG, Yandle TG, Doughty RN et al. (1997) Immunoreactive amino-terminal pro-brain natriuretic peptide (NT-PROBNP): a new marker of cardiac impairment. Clin Endocrinol (Oxf) 47: 287-296. [Crossref]
- Codognotto M, Piccoli A, Zaninotto M, Mion M, Plebani M et al. (2007) Renal dysfunction is a confounder for plasma natriuretic peptides in detecting heart dysfunction in uremic and idiopathic dilated cardiomyopathies. Clin Chem 53: 2097-2104. [Crossref]
- Takami Y, Horio T, Iwashima Y, Takiuchi S, Kamide K et al. (2004) Diagnostic and prognostic value of plasma brain natriuretic peptide in non-dialysis-dependent CRF. Am J Kidney Dis 44: 420-428. [Crossref]
- Apple FS, Murakami MM, Pearce LA, Herzog CA (2004) Multi-biomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 50: 2279-2285. [Crossref]
- Wang AY, Lam CW, Yu CM, Wang M, Chan IH et al. (2007) N-terminal pro-brain natriuretic peptide: an independent risk predictor of cardiovascular congestion, mortality, and adverse cardiovascular outcomes in chronic peritoneal dialysis patients. J Am Soc Nephrol 18: 321-330. [Crossref]
- Mallamaci F, Zoccali C, Tripepi G, Benedetto FA, Parlongo S et al. (2001) Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 59: 1559-1566. [Crossref]
- Silberberg JS, Barre PE, Prichard SS, Sniderman AD (1989) Impact of left ventricular hypertrophy on survival in end-stage renal disease. Kidney Int 36: 286-290. [Crossref]
- Levin A, Thompson CR, Ethier J, et al. (1999) Left ventricular mass index increase in early renal disease: impact of decline in haemoglobin. Am J Kidney Dis 34: 125-134. [Crossref]
- London GM, Pannier B, Guerin AP, et al. (2001) Alterations of left ventricular hypertrophy in and survival of patients receiving haemodialysis: follow-up of an interventional study. J Am Soc Nephrol 12: 2759-2767. [Crossref]
- Zoccali C, Benedetto FA, Tripepi G, et al. (2001) Creed Investigators: cardiac natriuretic peptides are related to left ventricular mass and function and predict mortality in dialysis patients. J Am Soc Nephrol 12: 1508-1515. [Crossref]
- Yu WC, Lin IF, Chuang SY, Chen CH (2006) Effect of ramipril on left ventricular mass in normotensive hemodialysis patients. Am J Kidney Dis 47: 478-484. [Crossref]
- Cowan BR, Young AA (2009) Left ventricular hypertrophy and renin-angiotensin system blockade. Curr Hypertens Rep 11: 167-172. [Crossref]
- Burns J, Ball SG, Worthy G, et al. (2012) Hypertensive left ventricular hypertrophy: a mechanistic approach to optimizing regression assessed by cardiovascular magnetic resonance. J Hypertens 30: 2039-2046. [Crossref]
- Wang AY, Sanderson JE (2011) Treatment of heart failure in long-term dialysis patients: a reappraisal. Am J Kidney Dis 57: 760-772. [Crossref]
- Hage FG, De Mattos AM, Khamash H, Mehta S, Warnock D et al. (2010) QT Prolongation Is an Independent Predictor of Mortality in End-Stage Renal Disease. Clin Cardiol 33: 361-366. [Crossref]
- De Bie MK, Koopman MG, Gaasbeek A, Dekker FW, Maan AC et al. (2012) Incremental prognostic value of an abnormal baseline spatial QRS-T angle in chronic dialysis patients. Europace 15: 290-296. [Crossref]
- Genovesi S, Rossi E, Nava M, Riva H, De Franceschi S et al. (2013) A case series of chronic haemodialysis patients: mortality, sudden death, and QT interval. Europace 15: 1025-1033. [Crossref]
- Tereshchenko LG, Oehler A, Meoni L, Ghafoori E, Kim E et al. (2016) Electrophysiological Substrate and Risk of Mortality in Incident Hemodialysis Patients: Predictors of Arrhythmic and Cardiovascular Risk in End-Stage Renal Disease (PACE) Study. J Am Soc Nephrol 27: 3413-3420 [Crossref]
- Shafi S, Saleem M, Anjum R, Abdullah W, Shafi T (2017) ECG abnormalities in patients with chronic kidney disease. J Ayub Med Coll Abbottabad 29: 61-64. [Crossref]
- Stewart GA, Gansevoort RO, Mark PB, Rooney E, Mcdonagh TA et al. (2005) Electrocardiographic abnormalities and uremic cardiomyopathy. Kidney Int 67: 217-226. [Crossref]
- Stewart GA, Foster J, Cowan M, Rooney E, Mcdonagh T et al. (1999) Echocardiography overestimates left ventricular mass in haemodialysis patients relative to magnetic resonance imaging. Kidney Int 56: 2248-2253. [Crossref]
- Mark PB, Johnston N, Groenning BA, Foster JE, Blyth KG et al. (2006) Redefinition of uremic cardiomyopathy by contrast-enhanced cardiac magnetic resonance imaging. Kidney Int 69: 1839-1845. [Crossref]
- Patel RK, Oliver S, Mark PB, Powell JR, McQuarrie EP et al. (2009) Determinants of left ventricular mass and hypertrophy in hemodialysis patients assessed by cardiac magnetic resonance imaging. Clin J Am Soc Nephrol 4: 1477-1483. [Crossref]
- Ebeid AM, Elsharkawy EM, Fawal SK, Naga YS, Oraby M (2017) Role of cardiac magnetic resonance imaging and echocardiography in assessing the left ventricle in haemodialysis patients. Journal of the Egyptian Society of Nephrology and Transplantation 17: 30-37
- Rutherford E, Mangion K, McComb C, Bell E, Cockburn S et al. (2017) Myocardial changes in incident haemodialysis patients over 6-months: an observational cardiac magnetic resonance imaging study. Sci Rep 7: 13976. [Crossref]
- Gong IY, Al-Amro B, Prasad GR, Connelly PW, Wald RM et al. (2018) Cardiovascular magnetic resonance left ventricular strain in end-stage renal disease patients after kidney transplantation. J Cardiovasc Magn Reson 20: 83. [Crossref]
- Pecoits-Filho R, Barberato SH (2010) Echocardiography in chronic kidney disease: diagnostic and prognostic implications. Nephron Clin Pract 114: c242-c247. [Crossref]
- Altekin RK, Kucuk M, Yanikoglu A, Karakas MS, Er A et al. (2012) Evaluation of the left ventricular regional function using two-dimensional speckle tracking echocardiography in patients with end-stage renal disease with preserved left ventricular ejection fraction. Acta cardiologica 67: 681-691. [Crossref]
- Chen R, Wu X, Shen LJ, Wang B, Ma MM et al. (2014) Left ventricular myocardial function in hemodialysis and nondialysis uremia patients: a three-dimensional speckle-tracking echocardiography study. PloS One 9: e100265. [Crossref]
- Kramann R, Erpenbeck J, Schneider RK, Röhl AB, Hein M et al. (2014) Speckle tracking echocardiography detects uremic cardiomyopathy early and predicts cardiovascular mortality in ESRD. J Am Soc Nephrol 25: 2351-2365. [Crossref]
- Panoulas VF, Sulemane S, Konstantinou K, Bratsas A, Elliott SJ et al. (2014) Early detection of subclinical left ventricular myocardial dysfunction in patients with chronic kidney disease. Eur Heart J Cardiovasc Imaging 16: 539-548. [Crossref]
- Krishnasamy R, Isbel NM, Hawley CM, Pascoe EM, Burrage M et al. (2015) Left ventricular global longitudinal strain (GLS) is a superior predictor of all-cause and cardiovascular mortality when compared to ejection fraction in advanced chronic kidney disease. PLoS One 10: e0127044. [Crossref]
- Ali HA, Mohamad NA, Mohammad HS, Hamid SK (2016) 2-D Speckle tracking in the assessment of left and right ventricular functions in hemodialysis versus recently diagnosed uremic patients with preserved systolic function. The Egyptian Journal of Critical Care Medicine 4: 139-144.
- Ibrahim SS, Koura MA, Emara AA, Kamel M, El-Wahed WA (2016) The effect of hemodialysis-induced preload changes on the left ventricular function: a speckle-tracking echocardiographic study. Menoufia Medical Journal 29: 406.
- Hassanin N, Alkemary A (2016) Early detection of subclinical uremic cardiomyopathy using two-dimensional speckle tracking echocardiography. Echocardiography 33: 527-536. [Crossref]
- Van Huis M, Schoenmaker NJ, Groothoff JW, van der Lee JH, van Dyk M et al. (2016) Impaired longitudinal deformation measured by speckle-tracking echocardiography in children with end-stage renal disease. Pediatr Nephrol 31: 1499-1508. [Crossref]
- Hensen LC, Goossens K, Delgado V, Rotmans JI, Jukema JW et al. (2017) Prognostic implications of left ventricular global longitudinal strain in predialysis and dialysis patients. Am J Cardiol 120: 500-504. [Crossref]
- Ma Y, Zhang B, Zhang Y, Dong Y, Zhang R (2018) Ultrasonic image analysis of longitudinal strain in uremic patients with preserved left ventricular ejection fraction. Biomed Eng Online 17: 112. [Crossref]
- Tamulenaite E, Zvirblyte R, Ereminiene R, Ziginskiene E, Ereminiene E (2018) Changes of left and right ventricle mechanics and function in patients with end-stage renal disease undergoing haemodialysis. Medicina 54: 87. [Crossref]
- Pecoits-Filho R, Barberato SH (2010) Echocardiography in chronic kidney disease: diagnostic and prognostic implications. Nephron Clin Pract 114: c242-247. [Crossref]
- Liu YW, Su CT, Song EJ, Tsai WC, Li YH et al. (2015) The role of echocardiographic study in patients with chronic kidney disease. J Formos Med Assoc 114: 797-805. [Crossref]
- Waks JW, Tereshchenko LG, Parekh RS (2016) Electrocardiographic predictors of mortality and sudden cardiac death in patients with end stage renal disease on haemodialysis. J Electrocardiol 49: 848-854. [Crossref]
- Lima MS, Villarraga HR, Abduch MC, Lima MF, Cruz CB etal. (2017) Global Longitudinal Strain or Left Ventricular Twist and Torsion? Which Correlates Best with Ejection Fraction? Arq Bras Cardiol 109: 23-29. [Crossref]
- Langeland S, D’Hooge J, Wouters PF, Leather HA, Claus P et al. (2005) Experimental validation of a new ultrasound method for the simultaneous assessment of radial and longitudinal myocardial deformation independent of insonation angle. Circulation 112: 2157-2162. [Crossref]
- Helle-Valle T, Crosby J, Edvardsen T, Lyseggen E, Amundsen BH et al. (2005) New noninvasive method for assessment of left ventricular rotation - Speckle tracking echocardiography. Circulation 112: 3149-3156. [Crossref]
- Amundsen BH, Helle-Valle T, Edvardsen T, Torp H, Crosby J et al. (2006) Noninvasive myocardial strain measurement by speckle tracking echocardiography - Validation against sonomicrometry and tagged magnetic resonance imaging. J Am Coll Cardiol 47: 789-793. [Crossref]
- Sorrentino R, Esposito R, Pezzullo E, Galderisi M (2016) Real-time three-dimensional speckle tracking echocardiography: technical aspects and clinical applications. Research Reports in Clinical Cardiology 7: 147.
- Kis E, Ablonczy L, Reusz GS (2018) Cardiac magnetic resonance imaging of the myocardium in chronic kidney disease. Kidney and Blood Press Res 43: 134-142. [Crossref]
- Graham-Brown MP, Patel AS, Stensel DJ, March DS, Marsh AM et al. (2017) Imaging of myocardial fibrosis in patients with end-stage renal disease: current limitations and future possibilities. Biomed Res Int 2017: 5453606. [Crossref]