Ewing Sarcoma is a malign cancer that mainly occurs in white boys and that affects primitive mesenchymal cells. The main genetic alteration responsible for this cancer is EWSR1-FLI1 translocation that encodes a chimeric protein that can interfere in other genes transcription. This abnormality associated to secondary mutations generates the disease phenotype, affecting bones and soft tissues. For diagnosis purposes, it is necessary a histochemical and anatomopathological examination. Treatment is based on adjuvant chemotherapy associated to radiotherapy and surgery, with the intention to eradicate local neoplastic and metastatic cells, which normally stablish in the lungs.
ewing sarcoma, EWS-ETS, EWSR1-FLI1, bone cancer, osseous tumor
Ewing Sarcoma Family is a group of neoplasms with source on neuroectodermal primitive cells that have the potential to differentiate into various types of tumors including the Ewing sarcoma (ES), primitive neuroectodermal tumor (PNET), peripheral neuroepitelioma and Askin tumor [1]. ES and PNET are very associated, being the main difference between them the degree of cell dissemination, since in ES the cells are undifferentiated and in PNET they have a greater degree of differentiation [2-4]. In this review, the focus will be on the ES, specially its skeletal disease.
Genetically, ES is characterized by t(11;22)(q12;q11.2) translocation, one that fuses the EWSR1 gene on chromosome 22 with the FLI1 gene on chromosome 11, which is responsible for the tumorigenesis [4-6]. This chimeric oncogenic fusion encodes the EWS/FLI1 protein which is involved in many gene regulations and associated with secondary mutations, causing ES phenotype [5,7].
ES specifically initiates in the primitive mesenchymal stem cells of mesoderm or neural crest origin [5], and mainly affects bones (85%), specially pelvis, femur, tibia, chest wall and ribs [2,8,9], and in some cases soft tissues (15%)9. ES is the second most frequent primary skeletal tumor, only after osteosarcoma, and it has the most unfavorable prognosis. It is considered very aggressive and very rare, accounting with 0.2% of all cancers and 2.9% of the entire childhood one [2,10,11]. It has an annual incidence of 3: 1.000.000, affecting mainly children, adolescents and young adults, with a peak in the second decade of life [8,9,11,12]. 70% to 80% of patients are diagnosed younger than 20 years, 20% to 30% in the first decade and rarely before 5 years or after 30 years [2]. ES male/female rate is 1.5:1 approximately [2] with a significant association with ethic groups, being rare in African descendants and eastern Asia populations and more common in Hispanics [11,13,14].
Since this tumor belongs to a highly malignant cancer group it must be diagnosed early to begin treatment [11,12]. Moreover, it has a strong potential to metastasize, especially in the lungs, bone marrow and other bones. Many patients (20% to 25%) present metastasis at the initial diagnosis [2].
Therefore, the aim of this study is to revise the main genetic alterations associated to ES, its main symptoms and prognosis.
Most of the tumors occur due to structural aberrations in the chromosomes, such as translocations that involves the reciprocal exchange of DNA segments, usually without gain or loss of genetic material. Thus, chimeric chromosomes are formed [15]. Genetically, ES is characterized by the fusion of EWSR1 gene and members of the regulatory ETS genes, being the EWSR1-FLI1 junction the most frequent. This fusion occurs due to the translocation of the 11 and 22 chromosomes (t(11;22)(q24;q12)), which involves the gene FLI1 in the chromosome 11 that encodes a transcription factor related to cell proliferation, growth signs and tumorigenesis, and the gene EWSR1 in the chromosome 22 that encodes a multifunction protein involved in several cellular processes such as gene expression, cell signaling and RNA processing and transport [1,7,16].
In 10% to 15% of these translocations, the fusion occurs between the EWSR1 gene with other members of the regulatory ETS gene (such as t(21;22)(q22;q12)) that results in the hybrid gene EWSR1-ERG, or with other genes as ETV1, E1AF or FEV. In a much slow frequency, ES can be a result of the fusion of the RNA binding proteins TET family genes (TAF15 and TLS) with the members of the ETS family, resulting in chimeric TET-ETS genes that can encode oncoproteins [8]. Table 1 shows the main structural chromosome aberrations involved in ES.
Table 1. Structural chromosome aberrations in Ewing Sarcoma [15,17]
Diagnosis |
Chromosomal Aberrations |
Genes Involved |
Frequency (%) |
Ewing Sarcoma |
t(11;22)(q24;q12) |
EWS-FLI1 |
90 |
t(21;22)(q22;q12) |
EWS-ERG |
4 |
t(7;22)(p22;q12) |
EWS-ETV1 |
<1 |
t(17;22)(q12;q12) |
EWS-FEV |
<1 |
t(16;21)(p11;q22) |
FUS-ERG |
<1 |
The EWS-FLI1 gene fusion constitutively activates the promoter of the C-MYC gene, which is a proto oncogene that is essential in embryonic life and dependent on external and internal signals for its functioning. However, the C-MYC gene has reduced activity in differentiated cells and the inappropriate activation induces a gene amplification that results in the expression of proteins involved in the regulation of the cell cycle, differentiation and growth, and genomic instability that favors the appearance of neoplasms [18].
Chimeric EWS-FLI1 gene encodes proteins that can inhibit the apoptotic pathway and results in deregulated tumor growth [1,19]. These proteins can also activate the transcription of the TERT gene, regulating positively the telomerase expression, promoting immortality of the tumor cells [1,20].
ES has a very low mutational rate (0.15 mutations/Mb) compared to other malignant neoplasms. The most common secondary genetic lesions in ES are structural aberrations that include extra copy gain on chromosomes 8 and 12 and on the long arm of chromosome 1, and loss of copies on the long arm of chromosome 16 and on short arm of chromosome 9 [8].
The main somatic mutations in ES are in STAG2 (17%), CDKN2A (12%) and TP53 (7%) genes [12]. STAG2 encodes a complex of proteins known as Cohesive proteins that are involved with the chromosomal organization. This protein complex promotes cohesion among the chromatids during DNA duplication and presents a regulatory role during DNA transcription and DNA repair. Therefore, one deletion in STAG2 gene interferes in the chromosome segregation during mitosis, ending in accumulation of structural aberrations and aneuploidies [8,9,12].
CDKN2A is a tumor suppressor gene that regulates cell cycle by encoding proteins that can inhibit cyclin-dependent kinases (CDKs). CDKs activate the family of retinoblastoma proteins (pRB) that connect to transcription factors (E2F), blocking the cell cycle progression. In this way, one mutation on CDKN2A gene inactivates pRB that releases E2F, eliciting cell cycle progression [8,9,12].
TP53 is also a tumor suppressor gene that promotes DNA repair and can interrupt the cell cycle once activated. This tumor suppressor gene encodes the p53 protein that induces the synthesis of p21 protein that can inhibit the activity of the cyclin-CDK complexes stopping G1-S progression, interrupting the cell cycle. Therefore, one mutation on TP53 favors tumor growth, since the cell cycle will not be stopped. Thus, the constitutive activation of C-MYC gene by gene fusion EWS-ETS associated to mutations in secondary genes have a direct impact on tumor growth. All tumors that present mutations on STAG2 e TP53 genes are associated to aggressive tumors that present poor prognosis to conventional treatments and require alternative therapies [2,8,9,12].
EWS-FLI1 fusion promotes the remodeling of the chromatin, activating or repressing the expression of target genes through GGAA microsatellites. This means that the chimeric protein encoded by this fusion interacts with microsatellites with GGAA repetitive sequences, recruiting p300 protein, an acetyltransferase, that results in histone acetylation and chromatin opening, activating the enhancers and stimulating the target genes transcription, specially NR0B1, CAV1 and ERG2 genes [5] (Figure 1).
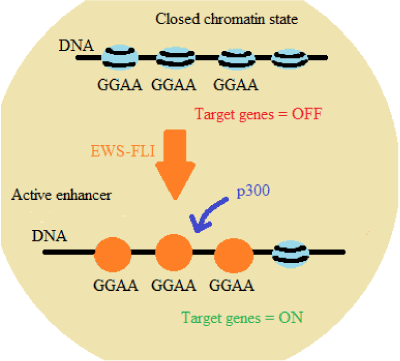
Figure 1: EWS-FLI1 interaction with microsatellites with GGAA repetitive sequences [5]
NR0B1 gene is responsible for DAX1 coding, which plays an important role on the production and function of several endocrine hormones and controls the activity of some genes during embryonic development. The aberrant expression of NR0B1 gene maintains the oncogenic transformation in ES cells. CAV1 gene expresses the caveolin-1 protein that is involved with endocytosis, being responsible for maintaining the cellular structure and regulating chemical signaling pathways. The imbalance of this gene expression is associated with tumor metastasis. ERG2 gene encodes the early growth response 2 protein, a transcription regulatory protein that activates several genes involved in the formation and maintenance of myelin, and is also associated with the proliferation of osteoprogenitor cells derived from mesoderm. Therefore, the activation of the GGAA repetitive sequence regions is a specific ability of the oncogenic protein expressed in ES and this activation regulates genes involved in tumorigenesis [5,21-23].
Changes in normal biological processes, such as duplication, transcription and translation, which are essential for normal embryonic development, are key features in determining tumor cell malignancy. And the embryonic cells differentiation is regulated by epigenetic mechanisms that are disrupted by the chimeric protein encoded by the EWS-FLI1 genes fusion. This protein changes the epigenetic regulation of homeobox (HOX) genes that are responsible for determining the pattern of the embryonic development, resulting in an abnormal expression of these genes in ES. The abnormal regulation of the HOX genes causes an erroneous migration of the neural crest cells and maintains the tumorous cells in an undifferentiated state [24].
EWS-FLI1 oncogenic fusion is also responsible for promoting the malignant transformation of the tumor, since it activates IGF-1 gene that is responsible for the extra and intracellular signal transduction that stimulates proliferation, growth and cell survival. Besides, IGF-1 is an important regulator of cell invasion and consequently of the metastasis in ES [25,26].
The earliest symptom in ES is acute pain that rapidly progresses and becomes chronic pain, strong enough to put the patient in analgesic drugs that sometimes does not help. Moreover, other symptoms can be noticed: fever, anemia, inflammation, moderate leukocytosis, increase in serum LDH, loss of appetite and weight, sometimes paresthesia and visible swelling in the site of the tumor can be noted [2].
The tumor is usually diagnosed by imaging [27]. First, in 2D radiograph, osteolysis and periostal reactions are discovered and magnetic resonance imaging (MRI) is more precise and reveals the extent of the tumor including intramedullary portion and soft tissue masses. MRI is also used for staging and surgical planning, and to evaluate therapeutic responses. However, definitive diagnosis depends on biopsy: sampling is more appropriate, but aspiration or core needle can also be used [2]. The biopsy is very important since many neoplasms present the same imaging characteristics as ES, e.g. avenocellular carcinoma, neuroblastoma, rhabdomyosarcoma, mesenchymal chondrosarcoma and lymphoblastic lymphoma.
Thus, anatomopathological, cytogenetic and immunohistochemical evaluations are essential to differentiate ES from other tumors [28,29]. Anatomopathological examination usually shows several irregular fragments of light brown coloring tissues and firm consistency. Cytogenetic studies in this type of tumor usually show chromosome aberrations. Immunohistochemical staining in ES normally is reported positive for CD99 (a cell surface glycoprotein responsible for cell adhesion), CD117 (a growth factor), CD56 (a neural cell adhesion molecule), and Ki67 (associated with cell proliferation) [1,2,28-30].
ES has an important metastatic potential, since 20% to 25% of ES patients have disseminated disease when they first seek for a doctor. This way, it is very important that every person diagnosed with ES is scanned for metastatic cells which mainly affect lungs, bone marrow and other bones [9,28].
ES is a very malignant tumor with poor prognosis. The overall survival for patients with localized ES that have undergone intensive treatment is 65% to 75% over a period of 5 years. Patients with metastasis have a survival rate of 30%, and for isolated and resectable lung metastasis the rate is 50%. The rate survival and prognosis for those with recurrent disease are the worst [9].
Infants, adolescent patients under 15 years and woman have a better prognosis. The prognosis becomes poorer when the tumor diameter is bigger than 8 cm and with a volume that exceeds 200 ml. After chemotherapy, the main factor to improve rate survival is how much the tumor could be resected and diminished [31,32]. Even for those in remission, the long-term effects of the cytotoxic treatment can be significative [31].
A better prognosis also depends on different factors. High levels of Ki67 is associated with a poor prognosis, while high levels of miR-34, a micro RNA responsible for controlling the cell cycle, proliferation and apoptosis, improve the prognosis [29]. High levels of LDH in the serum are also correlated to poor prognosis, a larger tumor and metastatic dissemination [32].
ES treatment is based on a multidisciplinary approach which increases the chances of cure. In a systematic treatment form, the first step is chemotherapy to control both primary tumor and possible metastatic cells. The traditional cytotoxic active agents used are known as VAC, vincristine, actinomycin D and cyclophosphamide, normally combined with doxorubicin, iofosfamide and etoposide. Common side effects of this treatment are nausea, vomiting, diarrhea, loss of appetite, loss of hair, weakness, bleeding, infections and infertility [9,12,29,33,34].
After chemotherapy, surgery and/or radiotherapy are used as local control therapy. The recommended dose for neoadjuvant radiotherapy is 54 to 55 gy with a safety margin of 2 cm. As sole treatment, radiotherapy results in a high risk of recurrence (30%-35%). In surgery, tumor resection is more recommended and amputation is rarely required. Often after all these therapies the patient receives additional chemotherapy in order to eliminate any residual abnormal cells [2,9,35].
New studies have been proposing different therapies for ES patients. It was discovered that the EWS-FLI junction is bonded with RNA helicase A, which plays an important role in the regulation of genetic transcription. This connection can be interfered by a small molecule, YK-4-279, that can destroy tumor cells and reduce tumor growth. ES is also very sensitive to PARP1 inhibitors, since PARP1 acts with BRCA1 and 2 at the DNA repair, connected to differentiation, proliferation and tumor transformation [33,36].
Iwamoto et al. [2] shows that Flavopiridol, a kinase inhibitor, could stop efficiently ES cells growth and inducing apoptosis. This research shows that therapies that could interfere in the cyclin-CDK complex could be very effective to stop tumor growth and could be a substantial hope to those who are affected by this disease.
Since ES is a very aggressive type of tumor and normally with poor prognosis, to understand all the genetic and molecular modifications involved with this cancer is essential for the development of new therapies and treatments that can somehow improve the patient prognosis.
The authors would like to acknowledge Medical School of Presidente Prudente for supporting this review.
- Zagar TM, Triche TJ, Kinsella TJ (2008) Ex2021 Copyright OAT. All rights reservyears later. J Clin Oncol 26: 4230-4232. [Crossref]
- Iwamoto Y (2007) Diagnosis and treatment of Ewing's sarcoma. Jpn J Clin Oncol 37: 79-89. [Crossref]
- Ushigome S, Machinami R, Sorensen PH (2002) Ewing sarcoma/primitive neuroectodermal tumour (PNET). In: Fletcher CDM, Unni KK, Mertens F (Edtr). World Health Organization Classification of Tumours. Pathology & Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press: 298-300.
- Li X, Tanaka K, Nakatani F, Matsunobu T, Sakimura R, et al. (2005) Transactivation of cyclin E gene by EWS-Fli1 and antitumor effects of cyclin dependent kinase inhibitor on Ewing’s family tumor cells. Int J Cancer 116: 385-394. [Crossref]
- Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, et al. (2014) EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 26: 668-681. [Crossref]
- Martinelli M, Parra A, Scapoli L, De Sanctis P, Chiadini V, et al. (2016) CD99 polymorphisms significantly influence the probability to develop Ewing sarcoma in earlier age and patient disease progression. Oncotarget 7: 77958-77967. [Crossref]
- Sannino G, Orth MF, Grünewald TG (2017) Next steps in Ewing sarcoma (epi-)genomics. Future Oncol 13: 1207-1211. [Crossref]
- Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, et al. (2014) Genomic Landscape of Ewing Sarcoma Defines an Aggressive Subtype with Co-Association of STAG2 and TP53 Mutations. Cancer Discov 4: 1342-1353. [Crossref]
- Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, et al. (2015) Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol 33: 3036-3046. [Crossref]
- Pillay Y, Ferreira N, Marais LC (2016) Primary malignant bone tumours: Epidemiological data from an Orthopaedic Oncology Unit in South Africa. SA Orthop J 15: 12-16.
- Baptista AM, Zumárraga JP, Dos Santos RP, Haubert GO, de Camargo OP (2016) SERUM VALUES OF ALKALINE PHOSPHATASE AND LACTATE DEHYDROGENASE IN EWING'S SARCOMA. Acta Ortop Bras24: 196-199. [Crossref]
- Brohl AS, Solomon DA, Chang W, Wang J, Song Y, et al. (2014) The Genomic Landscape of the Ewing Sarcoma Family of Tumors Reveals Recurrent STAG2 Mutation. PLoS Genet 10: e1004475. [Crossref]
- Parkin DM, Stiller CA, Nectoux J (1993) International variations in the incidence of childhood bone tumours. Int J Cancer 53: 371-376. [Crossref]
- Grier HE (1997) The Ewing family of tumors. Ewing's sarcoma and primitive neuroectodermal tumors. Pediatr Clin North Am 44: 991-1004. [Crossref]
- Ginsberg JP, de Alava E, Ladanyi M, Wexler LH, Kovar H, et al. (1999) EWS-FLI1 and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing's sarcoma. J Clin Oncol 17: 1809-1814. [Crossref]
- Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ (2005) Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res 65: 8984-8992. [Crossref]
- Grohar PJ, Janeway KA1, Mase LD1, Schiffman JD1 (2017) Advances in the Treatment of Pediatric Bone Sarcomas. Am Soc Clin Oncol Educ Book 37: 725-735. [Crossref]
- Bailly RA, Bosselut R, Zucman J, Cormier F, Delattre O, et al. (1994) DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. Mol Cell Biol 14: 3230-3241. [Crossref]
- Yi H, Fujimura Y, Ouchida M, Prasad DD, Rao VN, et al. (1997) Inhibition of apoptosis by normal and aberrant Fli-1 and erg proteins involved in human solid tumors and leukemias. Oncogene 14: 1259-1268. [Crossref]
- Takahashi A, Higashino F, Aoyagi M, Yoshida K, Itoh M, et al. (2003) EWS/ETS fusions activate telomerase in Ewing's tumors. Cancer Res 63: 8338-8344. [Crossref]
- Grunewald TGP, Bernard V, Gilardi-Hebenstreit P, Raynal V, Surdez D, et al. (2015) Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 47: 1073-1078. [Crossref]
- Beck R, Monument MJ, Watkins WS, Smith R, Boucher KM, et al. (2012) EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet 205: 304-312. [Crossref]
- Gangwal K, Sankar S, Hollenhorst PC, Kinsey M, Haroldsen SC, et al. (2008) Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proc Natl Acad Sci U S A 105: 10149-10154. [Crossref]
- Svoboda LK, Harris A, Bailey NJ, Schwentner R, Tomazou E, et al. (2014) Overexpression of HOX genes is prevalent in Ewing sarcoma and is associated with altered epigenetic regulation of developmental transcription programs. Epigenetics 9: 1613-1625. [Crossref]
- Sun H, Lin DC, Cao Q, Guo X3, Marijon H3, et al. (2016) CRM1 Inhibition Promotes Cytotoxicity in Ewing Sarcoma Cells by Repressing EWS-FLI1-Dependent IGF-1 Signaling. Cancer Res 76: 2687-2697. [Crossref]
- Ho AL, Schwartz GK (2011) Targeting of insulin-like growth factor type 1 receptor in Ewing sarcoma: unfulfilled promise or a promising beginning? J Clin Oncol 29: 4581-4583. [Crossref]
- Sá Neto JL de, Simão MN, Crema MD, Engel EE, Nogueira-Barbosa MH (2017) Diagnostic performance of magnetic resonance imaging in the assessment of periosteal reactions in bone sarcomas using conventional radiography as the reference. Radiol Bras 50: 176-181. [Crossref]
- Barzola-Navarro EJ, Flores-García JÁ, López-Guerra D, Tejera-Pérez C, Rodríguez-Díez N, et al. (2015) Duodenal Ewing´s sarcoma: Unusual location and atypical EWRS-1 translocation. Rev Esp Enferm Dig 107: 109-110. [Crossref]
- Marino MT, Grilli A, Baricordi C, Manara MC, Ventura S, et al. (2014) Prognostic significance of miR-34a in Ewing sarcoma is associated with cyclin D1 and ki-67 expression. Ann Oncol 25: 2080-2086. [Crossref]
- Evola FR, Costarella L, Pavone V, Caff G, Cannavò L, et al. (2017) Biomarkers of Osteosarcoma, Chondrosarcoma, and Ewing Sarcoma. Front Pharmacol 8: 150. [Crossref]
- Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, et al. (2014) The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4: 1326-1341. [Crossref]
- Jain S, Kapoor G (2010) Chemotherapy in Ewing's sarcoma. Indian J Orthop 44: 369-377. [Crossref]
- Khoury JD (2005) Ewing sarcoma family of tumors. Adv Anat Pathol 12: 212-220. [Crossref]
- Biermann JS, Chow W, Reed DR, Lucas D, Adkins DR, et al. (2017) NCCN Guidelines Insights: Bone Cancer, Version 2.2017. J Natl Compr Canc Netw 15: 155-167. [Crossref]
- Mirzaei L, Kaal SEJ, Schreuder HWB, Bartels RHMA (2015) The Neurological Compromised Spine Due to Ewing Sarcoma. What First: Surgery or Chemotherapy? Therapy, Survival, and Neurological Outcome of 15 Cases with Primary Ewing Sarcoma of the Vertebral Column. Neurosurgery 77: 718-725. [Crossref]
- Brenner JC, Feng FY, Han S, Patel S, Goyal SV, et al. (2012) PARP-1 inhibition as a targeted strategy to treat Ewing's sarcoma. Cancer Res 72: 1608-1613. [Crossref]