Amyotrophic lateral sclerosis (ALS), a systems neurodegeneration of adults, is typically progressive, fatal, and arises from death of lower and upper motorneurons (MN’s). We carried out RNA sequencing (RNA-seq) from cervical spinal cord MN’s isolated with laser capture microdissection (LCM) from ALS (n=4) and CTL (n=6) subjects. Cufflinks estimation of FPKM values (fragments per kilobase of exon per million reads), identified 3868 genes where the minimal mean FPKM was 2.0 in both groups. “Biologically blind” plots showed gene expression in ALS MN’s was increased 2 to 7-fold compared to CTL MN gene expression. Comparison of gene expression between ALS MN’s and CTL MN’s using false discovery rate (FDR) correction identified 263 genes increased in ALS with FDR<0.1. “Biologically smart” DAVID analyses revealed significant Gene Ontology (GO) groupings into vacuolar ATPase families. “Biologically blind” 3-D visualized gene expression networks (MiruÒ) showed separation into ALS and CTL MN networks. ALS consistently increases expression several fold in MN genes that significantly segregate into vacuolar ATPase families, implying increased recycling of cellular components in ALS MN’s. (172 words).
Amyotrophic lateral sclerosis, motor neurons, laser capture micro dissection, RNA sequencing, gene networks, vacuolar ATPase, autophagy
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive and usually fatal (in the absence of artificial ventilation) adult systems neurodegeneration that arises from premature, accelerated death of lower motor neurons (MN’s) in brainstem and spinal cord and upper MN’s in motor cortex [1]. The typical ALS sufferer develops muscle cramps, weakness, spasticity and muscle atrophy, complicated by dysphagia and ventilator insufficiency. Unless artificial ventilation is undertaken, death usually occurs 2-5 years after symptom onset.
A minority (~5-10%) of ALS occurs in families (fALS; [2]) and appears to arise from mutations in one of a group of at least 14 seemingly unrelated genes. The most studied of these fALS genes is SOD1 (superoxide dismutase-1, aka CuZn SOD, aka cytosolic SOD), with over 100 causal mutations known. The most frequently occurring mutated fALS gene is a hexanucleotide repeat in the promoter or first exon of orf72, located on Chromosome 9, thus C9orf72 [1,3-8]. Mutations in C9orf72 are frequently associated with fronto-temporal dementia and are found in ~40% of fALS and up to 11% of sporadic ALS (sALS).
The cause(s) of sALS are unclear. Multiple cellular abnormalities have been described in sALS tissues, including increased oxidative stress damage, increased inflammatory signaling, depressed mitochondrial OXPHOS activity and calcium signaling, altered autophagy, abnormal accumulation of aggregated (presumably damaged) proteins, and increased activation of cell death signaling [5,9-35]. It is not known if most sALS arise from a single “genesis” event that leads to multiple cell biological deficits, or whether there are multiple different molecular genesis causes of sALS across individuals.
Examination of postmortem ALS tissues may provide insights into ALS etiologies. In a recent RNA-sequencing study of ALS cervical spinal cord sections [36], we described an apparent increase in neuroinflammation signaling involving tumor necrosis factor-alpha (TNF-α) as a major upstream regulator. Because whole spinal cord is mostly astrocytes, it was unclear what gene expression changes occur in motor neurons that are selectively vulnerable in ALS. In that context, it was of interest that pharmacological inhibition of microglial activation altered the course of ALS in a transgenic mouse model [37].
In the present study we used laser capture microdissection (LCM) to isolate cervical spinal cord MN’s in both ALS and CTL samples. In a prior publication we showed that qPCR of mtDNA-encoded genes in LCM-captured MN’s revealed loss of respiratory (OXPHOS) gene expression [38]. In the present study, from RNA-seq we identified significantly expressed nuclear genome encoded genes with a false-discovery rate (FDR) correction approach for multiple comparisons. We found 263 genes significantly overexpressed in ALS MN’s that were in Gene Ontology (GO) families significantly representing vacuolar ATPase. We also found 2 to 7-fold higher gene expression in ALS MN’s and separate gene expression networks between ALS and CTL MN’s.
Our findings suggest that by the time a subject dies from ALS, there appears to be increased recycling of intracellular components using the proton pumping vacuolar ATPase system. We also found gene expression evidence suggesting autophagy activation and epithelial-mesenchymal transition (EMT) in ALS MN’s, which if confirmed, would represent the first description of this phenomenon (EMT) in adult, non-neoplastic, central nervous tissues.
Supplemental Table 1 shows the demographics and characteristics of the ALS and CTL samples reported in this study. Note that between 614-1319 individual neurons were collected by laser-capture microdissection (LCM) from each sample for this study. We have previously reported that these LCM-isolated cervical spinal motorneurons had substantially increased neuronal and reduced glial markers, compared to whole cervical spinal cord, and showed significant expression of choline acetyl transferase, the enzyme responsible for synthesis of acetylcholine, the major transmitter used by motor neurons [38].
Table 1. “Biologically smart” DAVID
Annotation Cluster 1 |
Enrichment Score: 2.68 |
|
|
|
Category |
Term |
Genes |
Fold Enrichment |
Benjamini |
GOTERM_BP_DIRECT |
GO:0090383~phagosome acidification |
ATP6V0C, ATP6V1C1, RAB7A, ATP6V0A1, ATP6V1G2, ATP6V1B2 |
15.0 |
0.020 |
GOTERM_BP_DIRECT |
GO:0015991~ATP hydrolysis coupled proton transport |
ATP6V0C, ATP6V1C1, ATP6AP1, ATP1A3, ATP6V0A1, ATP6V1B2 |
12.7 |
0.036 |
GOTERM_BP_DIRECT |
GO:0008286~insulin receptor signaling pathway |
ATP6V0C, ATP6V1C1, GRB2, ATP6AP1, RHOQ, ATP6V0A1, ATP6V1G2, ATP6V1B2 |
6.9 |
0.044 |
GOTERM_BP_DIRECT |
GO:0033572~transferrin transport |
ATP6V0C, ATP6V1C1, ATP6AP1, ATP6V0A1, ATP6V1G2, ATP6V1B2 |
11.6 |
0.037 |
GOTERM_MF_DIRECT |
GO:0046961~proton-transporting ATPase activity, rotational mechanism |
ATP6V0C, ATP6V1C1, ATP6AP1, ATP6V0A1, ATP6V1B2 |
13.3 |
0.030 |
|
|
|
|
|
Annotation Cluster 2 |
Enrichment Score: 2.47 |
|
|
|
Category |
Term |
Genes |
Fold Enrichment |
Benjamini |
GOTERM_CC_DIRECT |
GO:0043194~axon initial segment |
NRCAM, ANK3, SCN8A, BIN1 |
26.4 |
0.009 |
|
|
|
|
|
Annotation Cluster 3 |
Enrichment Score: 2.23 |
|
|
|
Category |
Term |
Genes |
Fold Enrichment |
Benjamini |
GOTERM_CC_DIRECT |
GO:0005875~microtubule associated complex |
KIF1B, MAPT, MAP1A, MAP2, MAP1B |
10.7 |
0.022 |
Supplemental Table 2 shows expression of all genes (minus duplicates) we found in our Cufflinks-based analysis of RNA-seq data from LCM-isolated MN’s in CTL and ALS cervical spinal cord samples. It also shows the ratios of mean FPKM of ALS/mean FPKM CTL.
We used several “biologically blind” or “biologically smart” systems biology approaches to our gene expression data of LCM-isolated MN’s from CTL and ALS spinal cords:
The first approach was a commonly used comparative statistical analysis of FPKM values in each group (t-test), with correction for multiple comparisons based on the false discovery rate (FDR) approach. Using a FDR of <10% (q < 0.10), we found (Supplemental Table 3) 263 genes that were significantly (FDR<10%) overexpressed in ALS LCM MN. Table 1 shows the “biologically smart” DAVID ((Database for Annotation, Visualization and Integrated Discovery; https://david.ncifcrf.gov/; version 6.8 (released Oct., 2016)) gene ontology (GO) family results from analysis of these genes, where we report only GO results with Benjamini-corrected p values of < 0.05. GO biological processes identified included multiple families that used vacuolar ATPase genes, one process involving axon outgrowth and one process involving microtubule stabilization.
In all cases the genes in ALS MN’s were increased compared to those in CTL MN’s (Figure 1A). Those genes of the vacuolar ATPase family are separately plotted in Figure 1B.
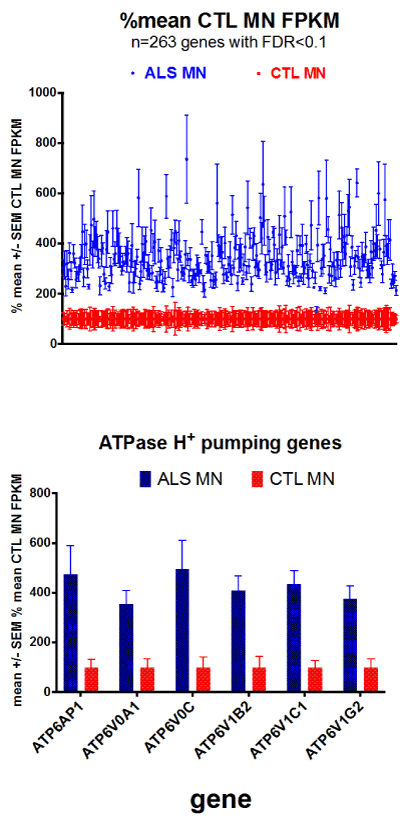
Figure 1 A. (top) Plots of ALS and CTL MN gene expression, where each sample was calculated as % of mean CTL FPKM, and means +/- SEM’s were plotted. B. (bottom) Bar chart plots of genes for vacuolar ATPase, plotted as means +/- SEM’s of % mean CTL values.
Second, we applied 3-D imaging of gene clusters; derived from Markov clustering of gene expression networks visualized using MiruÒ (www.kajeka.com). The .csv files used for Miru input are provided in Supplemental Table 4 for CTL and ALS MN’s, MiruÒ created 3-D networks from the 263 genes that were significantly (FDR < 0.1) overexpressed in ALS MN’s. Figure 2 shows the 3-D network created by MiruÒ for the ALS and CTL MNs’ gene expressions. The CTL and ALS MN’s were segregated into two separate groups. The one exception was inclusion of one CTL (CTL6) with the ALS MN group.
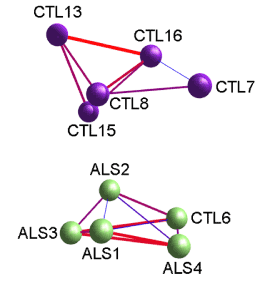
Figure 2. Screenshot from MiruÒ 3D network construction of ALS and CTL MN gene expression, after Markov clustering, of FPKM values for 263 genes that were significantly altered (FDR<0.1). Starting correlation coefficient was 0.9 (Pearson) and final clustering correlation coefficient was 0.95. Layout algorithm used was FMMM. Note that MiruÒ separated samples into two networks, one exclusively CTL (top), and one ALS (bottom) that had one CTL sample (CTL6).
In spite of its rarity and status as an orphan disease, ALS is devastating for those who have it. ALS is a cause of rapidly progressing disability and a premature, cruel death by ventilator insufficiency. Other than rare, familial genetic variants, causes of ALS and disease-altering therapies remain elusive.
Disruption of virtually every major cell function has been implicated in ALS etiology (see above). In our prior work using gene expression as a proxy for cell activity, we reported that inflammation, and in particular that mediated by TNF-α, in our cervical spinal cord specimens appeared to be playing an etiologic role [36], but inflammation was not the only process we found to be disturbed.
In the present study we isolated with laser capture micro dissection between ~600 and ~1300 anterior motor neurons per sample from the ALS and CTL cervical spinal cord sections available to us. We then isolated total RNA from these motor neurons, amplified it and subjected it to RNA-sequencing for an overview of all expressed genes. We used several systems biology approaches for analysis of this dataset that contained genes that were significantly altered (by FDR analysis) in ALS MN’s.
By using the publicly available DAVID [39,40] over-representation algorithm, we found that ALS motor neurons were significantly over-represented in gene ontology families involving vacuolar ATPase function.
Vacuolar ATPase (V-ATPase) is a 900 kDa multiple subunit, rotary ATPase complex that pump protons into multiple intracellular organelles [41,42]. Bafilomycin-A1, a V-ATPase inhibitor, induces an Atg-7 (autophagy protein)-dependent neuronal apoptosis [43], demonstrating one of the many important roles played by V-ATPase in the life of neurons. Although our observed increased expression in ALS MN’s of multiple autophagy genes and V-ATPase genes may be unrelated, it is tempting to speculate that one of the consequences in MN’s of ALS is increased autophagy.
Several autophagy-related genes were overexpressed in ALS MN’s. These included the autophagy-related proteins ATG1 (ALS/CTL=1.88); ATG13 (2.28); ATG2B (2.39); ATG3 (1.87); ATG9A (2.20); LAMP2 (3.28); and MAP1LC3B (LC3B) (3.84). These findings suggest that autophagy function in the ALS MN’s is increased and are consistent with the DAVID results.
Increased expression of LC3B has been noted in brainstems of G93A mutant SOD1 transgenic mice [44], and autophagy appears to be increased in motor neurons from sporadic ALS cases [45]. Autophagy is also abnormal in both sALS and fALS [46]. Additional autophagy-related genes overexpressed in LCM-MN’s from ALS subjects include DCTN1 (ALS/CTL=2.93) that encodes the p150 subunit of the transporter protein dynactin, and Rab7a (ALS/CTL= 3.98) that encodes the small GTPase Rab7 that regulates the maturation of autophagosomes, amphisomes, and late endosomes in cells [47]. In toto, the gene expression data are consistent with an over-activation of the autophagy system in ALS MN’s.
There is also the possibility that in ALS neurons is losing their integration into extracellular matrix, such as occurs in cancer metastasis during “epithelial-mesenchymal transition” (EMT). Recent studies in non-CNS cells have demonstrated that EMT can occur with activation of autophagy stimulated by reactive oxygen species [48]. We found that expression of two EMT markers were increased in ALS motor neurons, vimentin (ALS/CTL = 2.6) and N-cadherin (CDH2, ALS/CTL = 2.9). E-cadherin (CDH1) expression, normally reduced during EMT, was not detectable in ALS MN’s by RNA-seq. Other stresses may lead to the changes we observed, that may result in gene expression changes mimicking EMT.
Our post-mortem tissue study has many limitations. These include:
Limited sample numbers: We had only so many ALS and CTL cervical spinal cord samples available to us, from which we were able to extract motor neurons with high-enough RNA quality to be useful. In addition, it required almost a year for a skilled operator (ACL) to capture enough neurons from a total of 10 cervical spinal cord sections to yield RNA’s of sufficient qualities for amplification, carry out amplification and the qPCR studies. In addition, RNA-sequencing is costly, even though we carried out our own library preparation and bioinformatics.
Post-mortem status: All of our ALS subjects died from ALS; thus, they were at end-stage, and our findings may primarily represent survival strategies among neurons still present. Those neurons that have died may have other deficits. We have no insights into the temporal sequences of appearance of the deficits we appear to have uncovered, nor do we have insights into their origins.
Limitations of gene expression: Cells live and die usually by the actions of proteins, not RNA’s. Gene expression can provide a “snapshot” of what cells and tissues are experiencing and perhaps new directions they may utilize as adaptive strategies. However, gene expression studies don’t necessarily reflect the final status of proteins in cells, as other factors (particularly microRNA’s) can alter protein translation from mRNA genes. Thus, abnormalities in gene expression may or may not ultimately be reflected in cellular protein changes.
Cell heterogeneity: In order to carry out quantitative gene expression with current technology, it was necessary to isolate hundreds of motor neurons that could be pooled to yield RNA samples for amplification, multiplex library construction and sequencing. Thus, our findings represent at best population averages, and one should not assume that all neurons in the sample are behaving identically (they likely are not). Hopefully as technology advances, more single neuron RNA-seq studies can be carried out from tissue sections that would yield a more diverse picture of cellular responses to disease processes.
In spite of these significant limitations, we feel our results suggest additional directions for further investigations and experimental therapeutics. In terms of additional investigations, it might be worthwhile to pursue the possibility that the “prolonged autophagic death” described by Martin [49] and increased autophagy markers in sporadic ALS MN’s described by Sasaki [45] might arise as a result of oxidative stress-driven autophagy [48] through activation of epithelial-mesenchymal transition (EMT) of motor neurons. EMT is not known to occur normally in non-malignant tissues except during developmental neuronal polarization and migration [50], amniotic membrane rupture [51], organ fibrosis [52] and cardiac cells in culture [53]. EMT can be stimulated by a variety of factors, including TNF-α. Thus, it was of interest that two TNF receptor superfamilies (“SF”) showed increased expression in ALS MN’s: TNFRSF1A (ALS/CTL = 3.0) and TNFRSF21 (ALS/CTL = 2.0). The latter is associated with NF-kβ signaling, which has been shown to mediate EMT of non-neoplastic cardiac cells [53]. If EMT is occurring in adult ALS motor neurons, this would represent a new paradigm, amenable to inhibition, such as has been demonstrated with rapamycin treatment of cervical carcinoma cells [54] and curcumin treatment of human kidney tubular epithelial cells [55].
It would seem advantageous to treat sALS subjects for multiple simultaneous abnormalities. These could include strategies to detoxify free radicals and reduce oxidative stress, strategies to reduce inflammation in the CNS, and strategies to stimulate mitochondrial biogenesis so as to increase expression of respiratory (OXPHOS) proteins [56]. It is useful to consider the possibility that ALS subjects might be heterogeneous in these problems, with different subjects having different “proportions” of each deficit.
Our methods for acquisition of cervical spinal cord, isolation by LCM of motor neurons from cervical spinal cord sections, RNA isolation and amplification, cDNA generation and qPCR of mitochondrial genes have been previously described [26,38]. Methods for generation and quantitation of multiplex RNA-seq libraries from these amplified RNA’s have also been described [38]. Paired-end RNA sequencing of multiplex libraries made by one of us (DGB) were carried out by Cofactor Genomics (https://www.cofactorgenomics.com). Compressed sequencing files were examined by FastQC then trimmed of sequencing ends using Trimmomatic. Minimum Phred scores were 20 (sequencing accuracy = 99%). Trimmed sequences were aligned against the human hg38 genome using Tophat2/Bowtie2 and quantitated against the hg38 transcriptome using Cufflinks (JPB) as described in [36]. Statistics utilized version 7 of Prism for Mac (www.GraphPad.com).
Motor neurons from ALS subjects, isolated by laser capture microdissection (LCM) and analyzed by RNA sequencing against the hg38 version of the human genome, showed multiple gene expression abnormalities. The most prominent involved vacuolar ATPase systems, but other notable abnormalities included increased autophagy gene expression and markers of endothelial-mesenchymal transition. Interruption of these processes may reduce motor neuron death in ALS that morphologically is autophagic.
None of the authors has any conflicts of interest with the findings of this paper.
This research was supported by ALS Worldwide and the VCU Parkinson’s Research Center Funds, administered by the Medical College of Virginia Foundation. Neither of the Sponsors had any influence over data collection, analysis or decision to publish.
ACL carried out all tissue sectioning, LCM isolation of neurons, RNA extractions and amplifications. DGB performed all sequencing library construction and quantification. JPB designed the study, carried out the bioinformatics analyses and wrote the manuscript draft. All authors have seen and approved the final version.
- Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9: 617-628. [Crossref]
- Renton AE, Chiò A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17: 17-23. [Crossref]
- Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, et al. (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18: 1226-1229. [Crossref]
- Nordin A, Akimoto C, Wuolikainen A, Alstermark H, Jonsson P, et al. (2015) Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum Mol Genet 24: 3133-3142. [Crossref]
- Prudencio M, Belzil VV, Batra R,2021 Copyright OAT. All rights reservistinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nature neuroscience 18: 1175-1182. [Crossref]
- Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, et al. (2015) C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 14: 291-301. [Crossref]
- Satoh J, Yamamoto Y, Kitano S, Takitani M, Asahina N, et al. (2014) Molecular network analysis suggests a logical hypothesis for the pathological role of c9orf72 in amyotrophic lateral sclerosis/frontotemporal dementia. J Cent Nerv Syst Dis 6: 69-78. [Crossref]
- Xi Z, Zhang M, Bruni AC, Maletta RG, Colao R, et al. (2015) The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol 129: 715-727. [Crossref]
- Allen SP, Duffy LM, Shaw PJ, Grierson AJ (2015) Altered age-related changes in bioenergetic properties and mitochondrial morphology in fibroblasts from sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 36: 2893-2903. [Crossref]
- Andrus PK, Fleck TJ, Gurney ME, Hall ED (1998) Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. Journal of neurochemistry 71: 2041-2048. [Crossref]
- Babu GN, Kumar A, Chandra R, Puri SK, Kalita J, et al. (2008) Elevated inflammatory markers in a group of amyotrophic lateral sclerosis patients from northern India. Neurochemical research 33: 1145-1149. [Crossref]
- Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, et al. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137: 2329-2345. [Crossref]
- Chang Y, Kong Q, Shan X, Tian G, Ilieva H, et al. (2008) Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One 3: e2849. [Crossref]
- Cozzolino M, Rossi S, Mirra A, Carrì MT (2015) Mitochondrial dynamism and the pathogenesis of Amyotrophic Lateral Sclerosis. Front Cell Neurosci 9: 31. [Crossref]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, et al. (2006) Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. Journal of neurochemistry 96: 1349-1361. [Crossref]
- Danzeisen R, Schwalenstoecker B, Gillardon F, Buerger E, Krzykalla V, et al. (2006) Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2-amino-4,5,6,7-tetrahydro-6-Lpropylamino-benzathiazole dihydrochloride]. J Pharmacol Exp Ther 316: 189-199. [Crossref]
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, et al. (1997) Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. Journal of neurochemistry 69: 2064-2074. [Crossref]
- Finsterer J, Zarrouk-Mahjoub S (2016) Mitochondrial Disorders May Mimic Amyotrophic Lateral Sclerosis at Onset. Sultan Qaboos Univ Med J 16: e92-95. [Crossref]
- Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, et al. (2017) Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci Ther 23: 5-22. [Crossref]
- Hall ED, Andrus PK, Oostveen JA, Fleck TJ, Gurney ME (1998) Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. Journal of neuroscience research 53: 66-77. [Crossref]
- Higgins CM, Jung C, Xu Z (2003) ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci 4: 16. [Crossref]
- Jaiswal MK (2014) Selective vulnerability of motoneuron and perturbed mitochondrial calcium homeostasis in amyotrophic lateral sclerosis: implications for motoneurons specific calcium dysregulation. Mol Cell Ther 2: 26. [Crossref]
- Jiang Z, Wang W, Perry G, Zhu X, Wang X (2015) Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Transl Neurodegener 4: 14. [Crossref]
- Keeney PM, Bennett JP (2010) ALS spinal neurons show varied and reduced mtDNA gene copy numbers and increased mtDNA gene deletions. Molecular neurodegeneration 5: 21. [Crossref]
- Kong J, Xu Z (1998) Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. The Journal of neuroscience 18: 3241-3250. [Crossref]
- Ladd AC, Keeney PM, Govind MM, Bennett JP (2014) Mitochondrial oxidative phosphorylation transcriptome alterations in human amyotrophic lateral sclerosis spinal cord and blood. Neuromolecular Med 16: 714-726. [Crossref]
- Liu D, Wen J, Liu J, Li L (1999) The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB journal 13: 2318-2328. [Crossref]
- Malkki H (2015) Motor neuron disease: Brain transcriptome profiling reveals involvement of divergent pathways in C9orf72-associated and sporadic ALS. Nat Rev Neurol 11: 484. [Crossref]
- Martin LJ (2000) p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol Dis 7: 613-622. [Crossref]
- Martin LJ (2006) Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 65: 1103-1110. [Crossref]
- Martin LJ (2010) The mitochondrial permeability transition pore: a molecular target for amyotrophic lateral sclerosis therapy. Biochim Biophys Acta 1802: 186-197. [Crossref]
- Martin LJ (2011) Mitochondrial pathobiology in ALS. J Bioenerg Biomembr 43: 569-579. [Crossref]
- Sasaki S, Iwata M (2007) Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Journal of neuropathology and experimental neurology 66: 10-16. [Crossref]
- Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, et al. (2016) The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med 22: 869-878. [Crossref]
- Zhao W, Beers DR, Appel SH (2013) Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. Journal of neuroimmune pharmacology 8: 888-899. [Crossref]
- Brohawn DG, O'Brien LC, Bennett JP (2016) RNAseq Analyses Identify Tumor Necrosis Factor-Mediated Inflammation as a Major Abnormality in ALS Spinal Cord. PLoS One 11: e0160520. [Crossref]
- Trias E, Ibarburu S, Barreto-Núñez R, Babdor J, et al. (2016) Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J Neuroinflammation 13: 177. [Crossref]
- Ladd AC, Brohawn DG, Thomas RR, Keeney PM, Berr SS, et al. (2017) RNA-seq analyses reveal that cervical spinal cords and anterior motor neurons from amyotrophic lateral sclerosis subjects show reduced expression of mitochondrial DNA-encoded respiratory genes, and rhTFAM may correct this respiratory deficiency. Brain Res 1667: 74-83. [Crossref]
- Huang da W, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1-13. [Crossref]
- Huang da W, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44-57. [Crossref]
- Marshansky V, Rubinstein JL, Gruber G (2014) Eukaryotic V-ATPase: novel structural findings and functional insights. Biochim Biophys Acta 1837: 857-879. [Crossref]
- Rawson S, Harrison MA, Muench SP (2016) Rotating with the brakes on and other unresolved features of the vacuolar ATPase. Biochem Soc Trans 44: 851-855. [Crossref]
- Walls KC, Ghosh AP, Franklin AV, Klocke BJ, Ballestas M, et al. (2010) Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem 285: 10497-10507. [Crossref]
- An T, Shi P, Duan W, Zhang S, Yuan P, et al. (2014) Oxidative stress and autophagic alteration in brainstem of SOD1-G93A mouse model of ALS. Mol Neurobiol 49: 1435-1448. [Crossref]
- Sasaki S (2011) Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 70: 349-359. [Crossref]
- Webster CP, Smith EF, Shaw PJ, De Vos KJ (2017) Protein Homeostasis in Amyotrophic Lateral Sclerosis: Therapeutic Opportunities? Front Mol Neurosci 10: 123. [Crossref]
- Otomo A, Pan L, Hadano S (2012) Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases. Neurol Res Int 2012: 498428. [Crossref]
- Sioutas A, Vainikka LK, Kentson M, Dam-Larsen S, Wennerström U, et al. (2017) Oxidant-induced autophagy and ferritin degradation contribute to epithelial-mesenchymal transition through lysosomal iron. J Inflamm Res10: 29-39. [Crossref]
- Martin LJ (1999) Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol 58: 459-471. [Crossref]
- Seong E, Yuan L, Arikkath J (2015) Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adh Migr 9: 202-213. [Crossref]
- Janzen C, Sen S, Lei MY, Gagliardi de Assumpcao M, Challis J, et al. (2017) The Role of Epithelial to Mesenchymal Transition in Human Amniotic Membrane Rupture. J Clin Endocrinol Metab 102: 1261-1269. [Crossref]
- Dalla Pozza E, Forciniti S, Palmieri M, Dando I (2017) Secreted molecules inducing epithelial-to-mesenchymal transition in cancer development. Semin Cell Dev Biol. [Crossref]
- Clark CR, Robinson JY, Sanchez NS, Townsend TA, Arrieta JA, et al. (2016) Common pathways regulate Type III TGFbeta receptor-dependent cell invasion in epicardial and endocardial cells. Cell Signal 28: 688-698. [Crossref]
- Cheng KY, Hao M (2017) Mammalian Target of Rapamycin (mTOR) Regulates Transforming Growth Factor-β1 (TGF-β1)-Induced Epithelial-Mesenchymal Transition via Decreased Pyruvate Kinase M2 (PKM2) Expression in Cervical Cancer Cells. Med Sci Monit 23: 2017-2028. [Crossref]
- Zhu FQ, Chen MJ, Zhu M, Zhao RS, Qiu W, et al. (2017) Curcumin Suppresses Epithelial-Mesenchymal Transition of Renal Tubular Epithelial Cells through the Inhibition of Akt/mTOR Pathway. Biol Pharm Bull 40: 17-24. [Crossref]
- Smith EF, Shaw PJ, De Vos KJ (2017) The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. [Crossref]