This article illustrates the importance of melt curve analysis (MCA) in interpretation of mild nutrogenomic micro(mi)RNA expression data, by measuring the magnitude of the expression of key miRNA molecules in stool of healthy human adults as molecular markers, following the intake of Pomegranate juice (PGJ), functional fermented sobya (FS), rich in potential probiotic lactobacilli, or their combination. Total small RNA was isolated from stool of 25 volunteers before and following a three-week dietary intervention trial. Expression of 88 miRNA genes was evaluated using Qiagen's 96 well plate RT2 miRNA qPCR arrays. Employing parallel coordinates plots, there was no observed significant separation for the gene expression (Cq) values, using Roche 480® PCR LightCycler instrument used in this study, and none of the miRNAs showed significant statistical expression after controlling for the false discovery rate. On the other hand, melting temperature profiles produced during PCR amplification run, found seven significant genes (miR-184, miR-203, miR-373, miR-124, miR-96, miR-373 and miR-301a), which separated candidate miRNAs that could function as novel molecular markers of relevance to oxidative stress and immunoglobulin function, for the intake of polyphenol (PP)-rich, functional fermented foods rich in lactobacilli (FS), or their combination. We elaborate on these data and present a detailed review on use of melt curves for analyzing nutigenomic miRNA expression data, which initially appear to show no significant expressions, but are actually more subtle than this simplistic view, necessitating the understanding of the role of MCA for a comprehensive understanding of what the collective expression and MCA data collectively imply.
biomarkers, DNA, miRNA, RNA, fermented sobya, pomegranate, PCR, SNPs
Gene expression and its control by miRNAs
Cell's gene expression profile determines its function, phenotype and cells' response to external stimuli, and thus help elucidate various cellular functions, biochemical pathways and regulatory mechanisms [1]. Several gene expression profiling methods at the RNA level have emerged during past years and have been successfully applied to cancer research. Profiling by microarrays allows for the parallel quantification of thousands of genes from multiple samples simultaneously, using a single RNA preparation, and has become valuable because microarrays are convenient to use, do not require large-scale DNA sequencing, gives a clear idea of cells' physiological state, and is considered a comprehensive approach to characterize cancer molecularly, as seen in studies on colon cancer [1].
Control of gene expression has been studied by miRNA molecules, a small non-coding RNA molecule (18–24 nt long), involved in transcriptional and post-transcriptional regulation of gene expression by inhibiting gene translation. MiRNAs silence gene expression through inhibiting mRNA translation to protein, or by enhancing the degradation of mRNA. Since first reported in 1993 [2], the number of identified miRNAs in June 2014, version 14.0, the latest miRBase release (v20) [3] contains 24,521 miRNA loci from 206 species, processed to produce 30,424 mature miRNA products. MiRNAs are processed by RNA polymerase II to form a precursor step which is a long primary transcript. Pri-miR is converted to miRNA by sequential cutting with two enzymes belonging to a class of RNA III endonucleases, Drosha and Dicer. Drosha converts the long primary transcripts to ~70 nt long primary miRNAs (pri-miR), which migrate to the cytoplasm by Exportin 5, and converted to mature miRNA (~22 net) by Dicer [4]. Each miRNA may control multiple genes, and one or more miRNAs regulate a large proportion of human protein-coding genes, whereas each single gene may be regulated by multiple miRNAs [5]. MiRNAs inhibit gene expression through interaction with 3-untranslated regions (3 UTRs) of target mRNAs carrying complementary sequences [4,5].
Effect of antioxidant polyphenols --abundant in Mediterranean diets-- on gene expression unraveled by the availability of molecular biology techniques, reveals our adaptation to environmental changes [6]. Efforts to study the human transcriptome have collectively been applied to tissue, blood, and urine (i.e., normally sterile materials), as well as stool (a non-sterile medium). Extraction protocols that employ commercial reagents to obtain high-yield, reverse-transcribable (RT) RNA from human stool in studies performed on colon cancer have been reported [7,8].
Micro(mi)RNAs as biomarkers, and their roles in disease processes
A biomarker is believed to be a characteristic indicator of normal biological processes, pathogenic processes, or pharmacological responses to therapeutic interventions. In contrast, clinical endpoints are considered as variables representing a study subject's health from his/her perspective [9]. A variety of biomarkers exist today as surrogates to access clinical outcomes in diseases, predict the health of individuals, or improve drug development. An ideal biomarker should be safe and easily measured, is cost effective to follow up, is modifiable with treatment, and is consistent across genders and various ethnic groups. Because we never have a complete understanding of all processes affecting individual's health, biomarkers need to be constantly reevaluated for their relationship between surrogate endpoints and true clinical endpoints [10]. MiRNAs have been used herein as biomarkers for assessing the effect of intake of PP-rich or fermented foods on the expression of 88 miRNA genes known to influence cancer.
Disease modulation by nutrients
Cardiovascular diseases due to hypercholesterolemia are considered a risk factor for Chronic Heart Disease (CHD), and chronic degenerative diseases --caused wholly or partially by dietary patterns-- represent the most serious threat to public health [11,12]. Moreover, nearly one-third of all cancer deaths are due to poor nutrition, lack of physical activity, and obesity; and these risk factors account for nearly 80% of large intestine, breast, and prostate cancers. Chronic inflammation is considered a common factor that contributes to the development and progression of these illnesses, which are caused by and/or modified by diet [13].
Pomegranate juice (PGJ) and derived products are considered the richest sources of polyphenolic compounds, with positive implication on TC, LDL-C and TG plasma lipid profile [14]. Moreover, anthocyanin and ellagitannins pigments, mainly punicalagins, inhibit the activities of enzymes 3-hydroxy-3-methylglutaryl-CoA reductase and sterol O-acyltransferase, important in cholesterol metabolism [15]. Probiotic bacteria also contribute to lowering plasma hyper cholestrolemia due to the above mechanism, caused by the probiotic bile salt hydrolase (BSH) activity. This probiotic enzyme hydrolyses conjugates both glycodeoxycholic and taurodeoxycholic acids to hydrolysis products, inhibiting cholesterol absorption and decreasing reabsorption of bile acid [16].
Colonic microbiota is a central site for the metabolism of dietary PP and colonization of probiotic bacteria. A dietary intervention study with probiotic strains from three Lactobacillus species (L. acidophilus, L. casei and L. rhamnosus) given to healthy adults, showed that bacterial consumption caused the differential expression of from hundreds to thousands of genes in vivo in the human mucosa [17]. The interaction of PP with the gut microbiota influences the expression of some human genes (i.e., nutritional transcriptomics), which mediates mechanisms underlying their beneficial effects [17]. Similar in vivo mucosal transcriptome findings have been reported when adults were given the probiotic L. plantarum, illustrating how probiotics modulate human cellular pathways, and show remarkable similarity to responses obtained for certain bioactive molecules and drugs [18].
Participants
Study subjects were 25 healthy adults, 20 to 34 years old; exclusion were: absence of metabolic diseases, no use of medication for the last 6 weeks, and no signs of allergy or hypersensitivity to food or ingested material. Compliance with the supplementation in all subjects was satisfactory, as assessed daily, and. all subjects continued their habitual diets throughout the study. The research protocol was approved by the institution review board, and all subjects gave written consent prior to their participation in the study.
Design of the study
Figure 1 shows the design of the nutrigenomic randomized study. Estimated dietary intake was assessed by 3 repeated food records, one week before they were enrolled in the trial. The average portion size consumed, as well as composition data values from nutrient composition of the food were combined to assess average daily energy and nutrient intakes by the “nutrisurvey” software program. The characteristics of the voluntary subjects who were enrolled in the study, the mean daily energy intake, as well as selected macro nutrients are presented in Table 1.

Figure 1. Study design.
Table 1. Composition of the supplements
Parameter |
Unit |
Dietary supplements |
|
|
Control (Portion served) |
FS (Portion served) |
PGJ (Portion served) |
FS+PGJ (Portion served) |
Portion Size |
g |
– |
170 |
250 |
150+10 |
Total Solids |
g |
– |
40.01 |
17.75 |
48 |
Carbohydrate |
g |
– |
51.1 |
32.75 |
59 |
Dietary Fiber |
g |
– |
54 |
0.25 |
48.6 |
Energy |
kcal |
– |
263 |
135 |
290 |
Lactobacillus |
diverse cfu/serving size |
– |
– |
5.1 x 109 |
4.5 x 109 |
Yeast |
cfu/serving size |
– |
– |
2.77 x 1010 |
2.44 x 1010 |
Total PP |
mg*/portion g |
– |
– |
519.1±8.75 |
207.65±3.5 |
Antioxidant activity |
(AEAC)** |
– |
7.74±1.33 |
11.35±2.2 |
11.37±2.2 |
*mg galic acid equivalent (GAE), FS: Functional fermented sobya; PGJ: Pomegranate juice
**mmol ascorbic acid equivalent antioxidant activity [5]
Supplements
Pomegranate was obtained in bulk from the Obour Public Market, Cairo, Egypt. Pomegranate fruits were peeled and the juice was extracted using a laboratory pilot press (Braun, Germany). The juice was distributed in aliquots of 100 or 250 grams in air tight, light-proof polyethylene bottles, and frozen at −20˚C, where pomegranate polyphenols remained stable. Sour sobya, a fermented rice porridge containing per gram 3 × 107 cfu diverse lactic acid bacteria (LAB) and 1 × 107 cfu Sacharomyces cerivisiae. with added ingredients such as milk, sugar and grated coconut, was purchased twice a week from the retail market, and saved in the refrigerator. Sobya is fermented rice. Table 2 illustrates the proximate initial and final mean urinary polyphenols, plasma and urinary antioxidative activity, urinary thiobarbituric acid reactive species (TBARS), and erythrocytic glutathione-S-transferase (GST).
Table 2. Initial and final mean urinary polyphenols, plasma and urinary antioxidative activity, urinary TBARS and erythrocytic GST
Parameter |
Unit |
Control |
Sobya |
Pomegranate |
Sobya +Pomegranate |
|
|
Baseline |
Final |
Baseline |
Final |
Baseline |
Final |
Baseline |
Final |
|
|
X±SE |
X±SE p |
X±SE |
X±SE p |
X±SE |
X±SE p |
X±SE |
X±SE p |
|
|
|
|
|
|
|
|
|
|
Urinary polyphenol |
GAE/mg creat |
10.36±1.8 |
8.11±2.2 |
11.84±6.2 |
9.86±1.8 |
5.70±1.4 |
55.23±21.7 <0.05 |
10.40±3.2 |
21.62±7.3 |
Urinary antioxidant activity |
AEAC/mg creat* |
9.74±2.0 |
8.13±2.7 |
3.89±09 |
10.30±2.3 |
7.18±0.9 |
46.57±18.0 <0.05 |
10.90±2.4 |
20.25±3.9 |
Urinary TBARS |
ug/mg creat |
83.04±12.1 |
75.17±15.3 |
82.77±27.8 |
29.97±4.4 |
173.93±44.8 |
51.48±8.2 <0.05 |
157.70±47.8 |
40.62±8.3 |
Plasma antioxidant |
AEAC/1oo ml |
6.36±2.81 |
5.99±2.66 |
3.70±0.33 |
4.55±0.27 |
3.64±0.30 |
5.92±0.68 <0.05 |
2.78±0.11 |
4.49±0.58 |
E-GST activity |
IU/g Hb |
5.94±3.3 |
5.45±4.1 |
4.26±0.5 |
7.21±0.8 <0.05 |
4.73±1.0 |
8.34±1.0 |
4.56±1.0 |
6.90±1.0 |
X±SE: Mean ± Standard Error, *:mmol ascorbic acid equivalent antioxidant capacity/mg creatinine; GAE: gallic acid equivalent, Mean values are significantly different if the P-values are less than 0,05 (P<0.05) [5]
Stool collection and storage
Stool was obtained from the 25 healthy adults, twice at day 0 and three weeks after the dietary intervention. All stools were collected with sterile, disposable wood spatulas in clean containers, after stools were freshly passed, and then placed for storage into Nalgene screw top vials (Thermo Fisher Scientific, Inc., Palo Alto, CA, USA), each containing 2 ml of the preservative RNA later (Applied Biosystems/Ambion, Austin, TX, USA), which prevents the fragmentation of the fragile mRNA molecule [7], and vials were stored at – 70 °C until samples were ready for further analysis. Total small RNA, containing miRNAs, was extracted from all frozen samples at once, when ready, and there was no need to separate mRNA containing small miRNAs from total RNA, as small total RNA was suitable to make ss miRNA c-DNA.
Extraction of total small RNA
A procedure used for extracting small total RNA from stool was carried out using a guanidinium-based buffer, which comes with the RNeasy isolation Kit®, Qiagen, Valencia, CA, USA, as we have previously detailed [7]. DNase digestion was not carried out, as our earlier work demonstrated no difference in RNA yield or effect on RT-PCR after DNase digestion [7]. The time to purify aqueous RNA from all of the 25 frozen stool samples was ~ three hours. Small RNA concentrations were measured spectrophotometrically at λ 260 nm, 280 nm and 230nm, using a Nano-Drop spectrophotometer (Themo-Fischer Scientific). The integrity of total RNA was determined by an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Palo Alto, CA, USA) utilizing the RNA 6000 Nano LabChip®. RNA integrity number (RIN) was computed for each sample using instrument's software [7].
Preparation of ss-cDNA for molecular analysis
The RT2 miRNA First Strand Kit® from SABiosciences Corporation (Frederick, MD, USA) was employed for making a copy of ss-DNA in a 10.0 µl reverse transcription (RT) reaction, for each RNA samples in a sterile PCR tube, containing 100 ng total RNA, 1.0 µl miRNA RT primer & ERC mix, 2.0 /µl 5X miRNA RT buffer, 1.0 µl miRNA RT enzyme mix, 1.0 µl nucleotide mix and Rnase-free H2O to a final volume of 10.0 µl. The same amount of total RNA was used for each sample. Contents were gently mixed with a pipettor, followed by brief centrifugation. All tubes were then incubated for 2 hours at 37oC, followed by heating at 95oC for 5 minutes to degrade the RNA and inactivate the RT. All tubes were chilled on ice for 5 minutes, and 90 µl of Rnase-free H2O was added to each tube. Finished miRNA First Strand cDNA synthesis reactions were then stored overnight at -20oC [7].
Use of cancer RT2 miRNA PCR array 96-well plate to study miRNAs' expressions
We used a SABiosciences RT2 miRNA qPCR Array Plate System for Human (Qiagen) to analyze miRNA expression using real-time, reverse transcription PCR (RT-qPCR) as a sensitive and reliable quantitative method for miRNA expression analysis. The arrays employ a SYBR Green real-time PCR detection system, which has been optimized to analyze the expression of many mature miRNAs simultaneously. Each 96-well array plate contains a panel of primer sets for 88 relevant miRNA focused pathways (one universal primer and one gene-specific primer for each miRNA sequence), plus four housekeeping genes (Human SNORD 48, 47 and 44, and U6), and two RNA and two PCR quality controls. Duplicate RT Controls (RTC) to test the efficiency of the miRNA RT reaction, with a primer set that detects the template synthesized from the built in miRNA External RNA Control (ERC). There are duplicate RT controls (RTC) to test the efficiency of the miRNA RT process, with a primer set to detect the template synthesized from the kit's built-in miRNA External RNA Control (ERC). There is also duplicate positive PCR controls (PPC) to test the efficiency of the PCR process, using a per-dispensed artificial DNA sequence and the primer set that detects it. The two sets of duplicate control wells (RTC and PPC) also test for inter-well, intra-plate consistency. The human RT2 miRNA PCR Arrays reflect miRNA sequences annotated by the Sanger miRBase Release 14. Figure 2 shows the layout of the MAH-102F array.
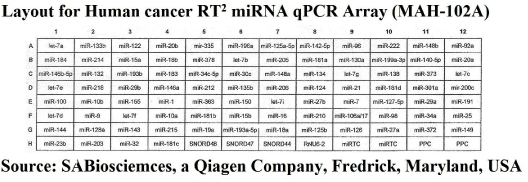
Figurge 2. Array layout.
Performing real-time quantitative polymerase chain reaction (qPCR)
We used RT2 SYBR Green qPCR Master Mix (SBA Biosciences) to obtain accurate results from our qPCR arrays. The following components were mixed in a 15-ml tube for 96-well plate format: 1275 µl of 2X RT2 SYBR Green PCR Master Mix, 100 µl of diluted first strand reaction, 1175 µl of ddH2O (total volume 2550 µl, of which 2400 was needed for 96 reactions, each well having 25 µl, with150 µl cocktail remaining.
We employed a Roche LightCycler 480® 96-well block PCR Machine (Roche, Mannheim, Germany) to carry out quantitative real-time miRNA expressions. When ready, we removed the needed miRNA qPCR Arrays, each wrapped in aluminum foil, from their sealed bags, added 25 µl of the same cocktail to each well, adjusted the ramp rate to 1oC/sec. We used 45 cycles in the program, and employed the Second Derivative Maximum method, available with the LightCycler 480® software for data analysis [19]. We first heated the 96 well plate for 10 min at 95oC to activate the HotStart DNA polymerase, then used a three-step cycling program (a 15 seconds heating at 95oC to separate the ds DNA, a 30 seconds annealing step at 60oC to detect and record SYBR Green fluorescence at each well during each cycle, and a final heating step for 30 seconds at 72oC). Each plate was visually inspected after the run for signs of evaporation from the wells. Data were analyzed using the 2-ΔΔCt method [20]. Resulting threshold cycle values for all wells were exported to a blank Excel sheet for analysis. We also ran a Dissociation (Melt) Curve Program after the cycling program [21] and generated a first derivative dissociation curve for each well in the plate, using the LC (Lightcycler's®) software.
Statistical and bioinformatics analysis
Gene expressions were standardized by dividing the SNORD48 value while raw melting temperatures were used. Analysis were done using the software R (version 3.1.3), with the package MASS [22]. One individual had so many missing values that this case was not used in the analysis so that the number of individuals is 24. For each standardized gene and each melting temperature, a one-way ANOVA was used to obtain a p-value. There were four levels of the explanatory variable: Control, Sobya, Pom, and Both. Parallel coordinate plots (parcoord command in R) [23] were used to visualize the data for each gene and each melting temperature. Coordinates were ordered using the magnitude of the p-value. The two sample t-test was used on gene expression to compare control to sobya and control to Both (t.test command in R with var.equal=FALSE). P-values were adjusted to control for false discovery rate. The method is outlined in [24] Benjamini and Yekutieli (p.adjust command in R with method=‘BY’).
We have bioinformatically correlated the 2-7 or 2-8 complement nucleotide bases in the mature miRNAs with the untranslated 3’ region of target mRNA (3’ UTR) of a message using a basic algorithm such as Broad’s Institute’s TargetScan [25] http://www.targetscan.org/archives.html, which provides a precompiled list for their prediction.
At base line, all participants in the trial excreted urinary total polyphenols; however, the inter individual variation was considerably high (4.89-12.59 mg GAE/100 ml urine).
Composition of the three supplements (FS, PGJ and FS + PGJ served to the volunteers is presented in Table 1. The initial and final mean urinary polyphenols, plasma and urinary antioxidative activity, urinary TBARS and erythrocytic GST, as well as the daily portion of PGJ provided 21 mg PP /day, and the combination of PGJ – FS was 9 mg PP /day, as presented in Table 2.
Figure 2 is a lay out of RT2 miRNA PCR Array Human Cancer microRNA (MAH-102A). Figure 6 is a graphical representation of the parallel plot coordinates of the studied miRNA genes for melting temperature curve analysis. The genes were ordered using the p-values of a one-way ANOVA based on groups. Genes with the smallest p-values are presented first. Figures 3 through 5 represent characteristics of melt curve analysis protocols.
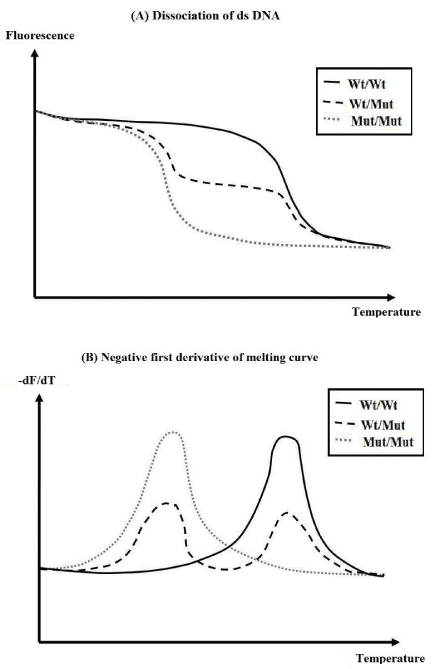
Figure 3. Thermophoresis of nucleic acids over a temperature gradient.
Figure 6a show eight employed control genes (Snord48, Snord47, Snord44, RNUU6-2. MiRTC1, miRTC2, and PPC1, PPC2). In Figure 6b, five miRNA genes (miR-184, miR-203, miR-124, miR-96 and miR-378) show clear separation. Gene miR-184 has the highest separation from the control gene. MiR-203 genes is hardly amplified in Sobya, while it is highly expressed in Pomegranate. For miR-373 gene, the control group is different from the other three treatment groups. For genes miR-124, miR-96 and miR-378, Pomegranate is well separated from other three groups. In Figure 6c, for gene miR-301a, the control is separated from the other three groups. Additional miRNA genes are not shown, as their p-values are greater (less significant), and the graphs did not show any meaningful separations.
Bioinformatics analysis using the TargetScan algorithm [25] for up-regulated and down-regulated mRNAs genes is shown in Table 3. The program yielded 21 mRNA genes encoding different cell regulatory functions. The first 12 of these mRNAs were found with the DAVID program [26] to be active in the nucleus and related to transcriptional control of gene regulation. For downregulated miRNAs, the DAVID algorithm found the first four of these mRNAs to be clustered in cell cycle regulation categories.
Table 3. Up-regulated and down-regulated target mRNA genes detected by a DAVID Bioinformatics algorithm
Up-regulated target mRNA genes
BCL11B, CUGBP2, EGR3, DLHAP2, NUFIP2, KLF3, MECP2, ZNF532, APPLI1, NFIB, SMAD7, SNF1LK, ANKRD52, C17orf39, FAM13A1, GLT8D3, KIAA0240, PCT, SOCS6, TNRC6B and UHRF1BP1 |
Down-regulated target mRNA genes
TGFB1, CKS2, IGF2, KLK10, FLNA, CSE1L, CXCL3, DPEP1 AND GUCA2B |
Suitability of stool as a medium for developing a sensitive molecular biomarker screen
Stool represents a challenging environment, as it contains many substances that may not be consistently removed in PCR, in addition to the existence of certain inhibitors, which all must be removed for a successful PCR reaction. Our results [27] and others [8,28] have shown that the presence of non-transformed RNA and other substances in stool do not interfere with measuring miRNA expressions, because of the use of suitable PCR primers, and the robustness of the real-time qPCR method [19]. Besides, stool colonocytes contain much more miRNA and mRNA than that available in free circulation, as in plasma [29], all factors that facilitate accurate and quantitative measurements.
PCR amplification and the effect of inhibitory substances
PCR has been used for miRNAs quantification because of its extreme sensitivity. This method, however, could lead to errors because of the presence of inhibiting substances, representing diverse compounds with different properties and mechanisms of action, which induce their effects by direct interaction with DNA that will be amplified, or through interference with the employed thermostable DNA polymerase [30].
Agents that reduce Mg2+ availability or interfere with the binding of Mg2+ to DNA polymerase could inhibit the PCR reaction [31]. Calcium ion is another inorganic inhibiting substance, although most PCR inhibitors are organic compounds (e.g., bile salts, sodium dodecyl sulphate, urea, phenol, ethanol, polysaccharides), as well as proteins (e.g., collagen, hemoglobin, immunoglobin G and protineases) [32]. The existence of polysaccharides in stool could decrease the capacity to resuspend precipitated RNA or disrupt the enzymatic reaction by mimicking the structure of nucleic acid. The DNA template of the PCR, as well as primers binding to DNA template can be inhibited by nucleases and other inhibitors, [33]. Remedial strategies for removal of inhibitors in stool, such as additional extraction steps, sephadex G-200 chromatography, heat treatment before the PCR, chloroform extraction, treatment with activated carbon, adding BSA, or dilution of sample, have been suggested [34]. We found the dilution method, in which the extracted ribonucleic acid (RNA) is diluted in the reaction mixture with distilled water or an isotonic buffer, to be the most practical method for preventing PCR inhibition using a commercially available diluent [35].
Role of biomarker miRNAs in various diseases
MiRNA functions were shown to regulate development [36] and apoptosis [37], and dysregulation of miRNAs has been associated with many diseases such as various cancers [38], heart diseases [39], kidney diseases [40]. nervous system diseases [41], alcoholism [42], obesity [43], auditory diseases [44], eye diseases [45], skeletal growth defects [46], as well as key role in host–virus pathogenesis of viral diseases [47]. A negative correlation was found between tissue specificity of interactions and miRNA in a number of diseases, and an association between miRNA conservation and disease, and predefined miRNA groups allow for identification of novel disease biomarkers at the miRNA level [48]. Specific miRNAs are crucial in oncogenesis [49], effective in classifying solid [50] and liquid tumors (51], and function as oncogenes or tumor suppressor genes [52]. MiRNA genes are often located at fragile sites, as well as minimal regions of loss of heterozygosity, or amplification of common breakpoints regions, suggesting their involvement in carcinogenesis [53]. MiRNAs have shown to serve as biomarkers for cancer diagnosis, prognosis and/or response to therapy [54,55]. Profiles of miRNA expression differ between normal tissues and tumor types, and evidence suggests that miRNA expression profiles can cluster similar tumor types together more accurately than expression profiles of protein-coding messenger (m)RNA genes [56]. Besides, small miRNAs (~18-22 nt long) are stable molecules than the fragile mRNA [27].
Melting curve analysis (MCA)
MCA is an assessment of dissociation characteristics of dsDNA during heating, leading to rise in absorbance, intensity and hyperchromicity. The temperature at which 50% of DNA is denatured is referred to as melting point, Tm.
Gathered information can be used to infer the presence of single nucleotide polymorphism (SNP), as well as clues to molecule's mode of interaction with DNA, such as intercalator slots in between base pairs through pi stacking and increasing salt concentration, leading to rise in melt temperature, whereas pH can affect DNA's stability, leading to lowering of its melting temperature [57]. Originally, strand dissociation was measured using UV absorbency, but now techniques based on fluorescence measurements using DNA intercalating fluorophores such as SYBR Green I, EvaGreen, or Fluorophore-labelled DNA probes (FRET probes) when they are bound to ds DNA [58] are now common. Specialized thermal cyclers that run the qPCR, such as Roche LightCycler(LC) 480®, used in this study, is programmed to produce the melt curve after the amplification cycles are completed. As the temperature increases, dsDNA denatures becoming ss and the dye dissociates, resulting in decrease in fluorescence. The graph of the negative first derivative of the melting-curve (-dF/dT) represents the rate of change of fluorescence in the amplification reaction and allows pin-pointing the temperature of dissociation (50% dissociation) using formed peaks to obviate or complement sequencing efforts [59].
The melting temperature (Tm) of each product is defined as the temperature at which the corresponding peak maximum occurs. The MCA confirms the specificity of the chosen primers, as well as reveals the presence of primer-dimers, which usually melt at lower temperatures than the desired product, because of their small size, and their presence severely reduce the amplification efficiency of the target gene as they compete for reaction components during amplification, and ultimately the accuracy of the data. The greatest effect is observed at the lowest concentrations of DNA, which ultimately compromises the dynamic range. Moreover, nonspecific amplifications may result in PCR products that melt at temperatures above or below that of the desired product. Optimizing reaction components (Mg2+, detergents, SYBR Green I concentration) and annealing temperatures aid in decreasing nonspecific product formation. Adequate product design, however, is considered to be the best method to avoid nonspecific products' formation. Including a negative control will determine if there is a coamplified genomic DNA [57,58]. The formula for Tm calculation is shown by the equation:
Tm = ____ ΣΔHon-n –-- 273.15 ,
ΣΔSon-n + RLnCT
where thermodynamic parameter ΔHo is Enthalpy changes, ΔSo parameter is Entropy changes, and CT is total strand concentration; these free-energy parameters predict Tm of most oligonucleotide duplexes to within 5oC; and permit prediction of DNA, as well as RNA duplex stabilities. It should be noted that Tm depends on the conditions of the experiment, such as oligonucleotide concentration, salts' concentration, mismatches and single nucleotide polymorphisms (SNPs) [60]. OligoAnalyzer® Tool [www.idtdna.com/analyzer/Applications/Oligoanalyzer] allows for calculating the Tm of employed nucleotides.
Microscale thermophoresis is a method that determines the stability, length, conformation and modifications of DNA and RNA. It relies on the directed movement of molecules in a temperature gradient that depends on surface characteristics of the molecule, such as size, charge and hydrophobicity. By measuring thermophoresis of nucleic acids over a temperature gradient, one finds clear melting transitions, and can resolve intermediate conformational states (Figure 3). These intermediate states are indicated by an additional peak in the thermophoretic signal preceding most melting transitions (Figure 3B) [59,61-63]. Agarose gel visualization is the gold standard for analyzing PCR products. Alternatively to reduce the number of gels needed to conform the presence of a single amplicon, “uMelt” melting curve prediction software (http://www.dna.utah/umelt/umelt.html) can be used to confirm that a single amplicon is generated by PCR [64]. This program predicts melt curves and their derivatives for qPCR-length amplicons and is suited to test for multiple peaks in a single amplicon product.
…Because SYBR Green I dye has several limitations, including inhibition of PCR, preferential binding to CG-rich sequences and effects on MCA, two intercalating dyes SYTO-13 and SYTO-82 were tried and did not show these negative effects, and SYTO-82 demonstrated a 50-fold lower detection limit [65], as well as best combinations of time-to threshold (Tt) and signal-to-noise ratio (SNR) [66]. To optimize performance of the buffer, a PCR mix supplemented with two additives, 1M 1,2-propanediol and 0.2 M trehalose, were shown to decrease Tm, efficiently neutralize PCR inhibitors, and increase the robustness and performance of qPCR with short amplicons [67]. “uAnalyzeSM“ is another web-based tool, similar to uMELT, for analyzing high-resolution melting PCR products' data, in which recursive nearest neighbor thermodynamic calculations are used to predict a melt curve. Using 14 amplicons of CYBB [cytochrome b-245 heavy chain, also known as cytochromae b (558) subunit], the main +/- standard deviation, the difference between experimental and predicted fluorescence at 50% helicity was -0.04 +/- 0.48oC [68].
MCA has been an effective and economical way for identification of virus stains [69], genes [70], bacterial strains [71], insect species [72], temperature validation of PCR cyclers [73], detection of translocations in lymphomas [74] and RNA interference/gene silencing [61]. Thus, the presence of double peaks during MCA, is not always indicative of non-specific amplification, and other methods such as agarose gel electrophoresis, and use of melt curve prediction software [61,68] are also needed in odder to determine the purity of an amplicon. For example, Figure 5A shows a single peak for exon 17b of CFTE (Cystic Fibrosis Transmembrane Conductance Regulator) gene, whereas the melt curve for an amplicon from exon 7 of CFTR shows two peaks, which could be interpreted as indicative of two separate amplicons (Figure 5B). However, analysis by agarose gel electrophoresis showed only one peak. To solve this conflict, an understanding of how melt curves are produced is needed. It should be emphasized that intercalating dyes used in qPCR, such as SYBR Green, will fluoresce only when the dye is bound to ds DNA, but not in the presence of a ssDNA, or when the DNA is free in solution. After the amplification cycle in qPCR, the instrument starts at a preset temperature above the primer Tm, and as the temperature increases dsDNA denatures becoming ssDNA and the dye therefore dissociates from the ssDNA (Figure 3A). The change in slope of this curve when blotted as a function of temperature to obtain a melt curve for CFTR exom 17b (Figure 4A). However, if we allow for the possibility that DNA my assume an intermediate state that is neither dsDNA or ssDNA, raw date from CFTR exon 7 melt will look Figure 4B. This could happen when there are regions of the amplicon that are more stable (e,g., G/C rich), which do not melt immediately, but maintain their ds configuration until the temperature becomes sufficiently high to melt it, which results in two phases (Figure 4B). Additional sequence factors, such as amplicon misalignment in A/T rich regions, and designs that have secondary structure in the amplicon region, can also produce products that melt in multiple phases.
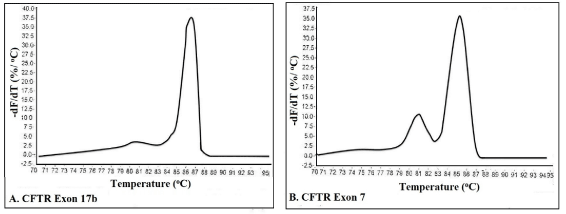
Figure 4. Melt curves for CFTR gene.
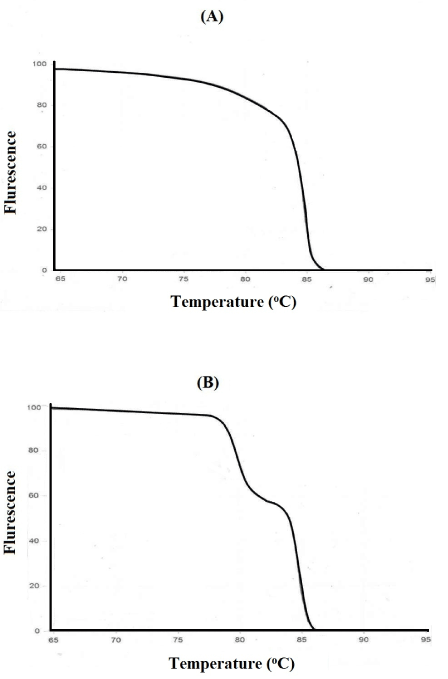
Figure 5. Analysis of cystic fibrosis transmembrane conductance regulator gene.
An advancement of MCA, referred to as High Resolution Melt (HRM), discovered and developed by Idaho Technology and the University of Utah [75, http://www.dna.utah.edu/Hi-Res/TOP_Hi-Res%20Melting.html], which has been useful for mutation detection and SNPs, enabling differentiation of homozygous wildtype, heterozygous and homozygous mutant alleles from the dissociation patterns. HRM has been used to identify variation in nucleic acid sequences, enabled by use of a more advanced software, and is therefore less expensive than probe-based genotyping methods, and allows for identification of variants quickly and accurately [76]. This method has been widely used in molecular diagnosis and for detection of mutations [77-79].
In our study, we found the melt curve analysis to be a useful and an informative method because after the statistical analysis carried on our miRNA expression samples showed no preferential expression of any of the 88 miRNA genes, a melt curve analysis on the same samples found that we could distinguish 7 miRNA (miR-184, miR-203, miR-373, miR-124, miR-96, miR-373 and miR-301a), due to different separation melting profiles (Figure 6). Thus, we believe that it is imperatives for investigators to run this kind of analysis on samples that particularly may not show expression differences in their mRNA or miRNA studied genes, such as nutritional samples.
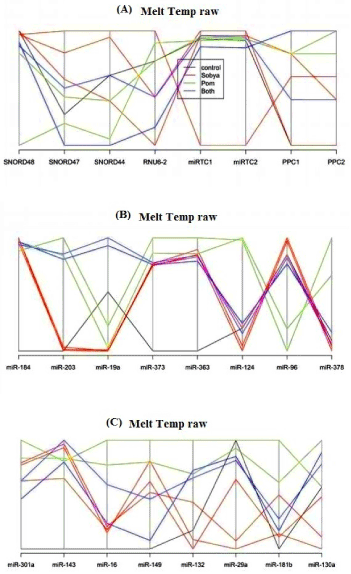
Figure 6. Graphical representation of parallel plot coordinates of the studies miRNA genes The genes were ordered using the p-values of a one-way ANOVA based on groups. Genes with the smallest p-values are presented first. Figure 1A below shows control genes, while in Figures 1B, and C, five miRNA genes show separation.
Bioinformatic methods to correlate seed miRNA Data with messenger(m)RNA Data
To provide information about complex regulatory elements, we correlated miRNA results with our available available mRNA data [80], as well as those data available in the open literature using computer model TargetScan [81,82]. The authenticity of functional miRNA/mRNA target pair once identified was validated by fulfilling four basic criteria: a) miRNA/mRNA target interaction can be verified, b) the predicted miRNA and mRNA target genes are co-expressed, c) a given miRNA must have a predictable effect on target protein expression, and d) miRNA-mediated regulation of target gene expression should equate to altered biological function. Bioinformatics showed 21 upregulated mRNA genes encoding different cell regulatory functions, and 12 of these mRNAs were found to be active in the nucleus and related to transcriptional control of gene regulation. For down-regulated miRNAs, four of the mRNAs appeared to be clustered in cell cycle regulation categories (Table 3) [27].
Clinical significance
2021 Copyright OAT. All rights reserv
The clinical significance of the study presented above is that using melting temperatures for analyzing nutrient-gene data is a promising new approach for identifying key regulatory miRNA genes related to metabolites rich in polyphenols, probiotic lactobacilli, or combinations of the two metabolites. Melt curve analysis is a powerful novel approach because after the statistical analysis carried on our miRNA samples produced negative gene expression (Cq) results, running melt curve analysis on the same samples identified 7 of the 88 miRNA genes imprinted on the highly sensitive focused PCR arrays (~ 8% of the genes), and using parallel coordinates plots showed noticeable separation of melt curve profiles. Thus, we believe that it is imperatives for investigators to run this kind of MCA on nutrition samples that are mild in nature, and many not always show significant differences in the expression of studied miRNA genes. The same analysis can also be envisioned for messenger mRNA amplifications, using mRNA arrays, and then using bioinformatics resources to correlate mRNA with miRNA data.
We are also planning to validate these initial results by carrying out additional miRNA nutrigenomic expression studies, with much more observations using PP, FS and their combinations, and collectively the obtained results would fully demonstrate the sensitivity/specificity of this powerful systemic molecular approach for analyzing nutrient-gene data.
We express our deep thanks to those volunteers who participated in this research by completing a dietary questionnaire, and providing urine, blood and stool samples for the study. We thank Dr, Clark D. Jeffries at Renaissance Computing Institute, University of North Carolina at Chapel Hill for his bioinformatics analysis, Funds for this research were provided by NIH Grant 1R43CA144823, and by GEM Tox Labs operating funds.
Authors certify that there has been no conflict of interest in carrying out this research, interpretation of data, or in preparing this manuscript for publication.
- Ahmed FE, Vos P, IJames S, Lysle DT, Allison RR, et al. (2007) Transcriptomic molecular markers for screening human colon cancer in stool and tissue. Cancer Genom Proteom 4: 1-20. [Crossref]
- Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843-854. [Crossref]
- Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68-73. [Crossref]
- Lund E, Dahlberg JE (2006) Substrate selectivity of exportin 5 and Dicer in the biogenesis of microRNAs. Cold Spring Harb Symp Quant Biol 71: 59-66. [Crossref]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, et al. (2005) Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769-773. [Crossref]
- Biomarkers Definition Working Group (2001) Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 69: 89-95. [Crossref]
- De Caterina R, Madonna R (2004) Nutrients and gene expression. World Rev Nutr Diet 93: 99-133. [Crossref]
- Link A, Balageur F, Shen Y, Nagasaka T, Lozano JJ, et al. (2010) Fecal MicroRNAs as novel biomarkers for colon cancer screening. Cancer Epidemiol Biomarkers Prev 9: 1766-1774. [Crossref]
- World Health Organization (WHO), Cardiovascular Diseases (CVDs), Who Fact Sheet No. 317, Geneva, 2011.
- Werner RM, Pearson TA (1998) LDL-cholesterol: a risk factor for coronary artery disease--from epidemiology to clinical trials. Can J Cardiol 14 Suppl B: 3B-10B. [Crossref]
- Benedetti S, Catalani S, Palma F, Canestrari F (2011) The antioxidant protection of CELLFOOD against oxidative damage in vitro. Food Chem Toxicol 49: 2292-2298. [Crossref]
- Bengmark S (2007) Advanced glycation and lipoxidation end products--amplifiers of inflammation: the role of food. JPEN J Parenter Enteral Nutr 31: 430-440. [Crossref]
- Gouda M, Moustafa A, Hussein L, Hamza M (2016) Three-week dietary intervention using apricots, pomegranate juice or/and fermented sour sobya and impact on biomarkers of antioxidative activity, oxidative stress and erythrocytic glutathione transferase activity among adults. Nutr J 15: 52. [Crossref]
- Esmaillzadeh A, Tahbaz F, Gaieni I, Alavi-Majd H, Azadbakht L (2006) Cholesterol-lowering effect of concentrated pomegranate juice consumption in type II diabetic patients with hyperlipidemia. Int J Vitam Nutr Res 76: 147-151. [Crossref]
- Zhuang G, Liu XM, Zhang QX, Tian F W, et al. (2012) Research advances with regards to clinical outcome and potential mechanisms of the cholestrol-lowering effects of probiotics. Clinical Lipidology 7: 501-507.
- van Baarlen P, Troost F, van der Meer C, Hooiveld G, Boekschoten M, et al. (2011) Human mucosal in vivo transcriptome responses to three lactobacilli indicate how probiotics may modulate human cellular pathways. Proc Natl Acad Sci U S A 108 Suppl 1: 4562-4569. [Crossref]
- Richards AL, Burns MB, Alazizi A, Barreiro LB, Pique-Regi R, et al. (2016) Genetic and transcriptional analysis of human host response to healthy gut microbiota. mSystems 1. [Crossref]
- Strimbu K, Tavel JA (2010) What are biomarkers? Curr Opin HIV AIDS 5: 463-466. [Crossref]
- Luu-The V, Paquet N, Calvo E, Cumps J (2002) Improved real-time RT-PCR measurements using second derivative calculation and double correction. BioTechniques 38: 287-293. [Crossref]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408. [Crossref]
- Ririe KM, Rasmussen RP and Wittwer CT (1997) Product determination by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 245: 154-160. [Crossref]
- Venables WN, Ripley BD (Eds.) (2002) Modern and Applied Stastistics. Fourth Edition. Springer, New York, USA.
- R Core Team R (2015) A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Australia.
- Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Annals Stat 28: 1165-1188.
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115: 787-798. [Crossref]
- Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44-57. [Crossref]
- Ahmed FE, Ahmed NC, Vos PW, Bonnerup C, Atkins JN, et al. (2013) Diagnostic microRNA markers to screen for sporadic human colon cancer in stool. I. Proof of principle. Cancer Genom Proteom 10: 93-114. [Crossref]
- Davidson LA, Lupton JR, Miskovsky E, Fields AP, Chapkin RS (2003) Quantification of human intestinal gene expression profiling using exfoliated colonocytes: a pilot study. Biomarkers 8: 51. [Crossref]
- Ahmed FE, Amed NC, Vos PW, Bonnerup C, Atkins JN, et al. (2012) Diagnostic microRNA markers to screen for sporadic human colon cancer in blood. Cancer Genom Proteom 9: 179-192. [Crossref]
- Abu Al-Soud W, Râdström P (1998) Capacity of nine thermostable DNA polymerases to mediate DNA amplification in the presence of PCR-inhibiting samples. Appl Environ Microbiol 64: 3748-3753. [Crossref]
- Wilson IG (1997) Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol 63: 3741-3751. [Crossref]
- Rådström P, Knutsson R, Wolffs P, Lövenklev M, Löfström C (2004) Pre-PCR processing: strategies to generate PCR-compatible samples. Mol Biotechnol 26: 133-146. [Crossref]
- Monteiro L, Bonnemaison D, Vekris A, Petry KG, Bonnet J, et al. (1997) Complex polysaccharides as PCR inhibitors in feces: Helicobacter pylori model. J Clin Microbiol 35: 995-998. [Crossref]
- Schrader C, Schielke A, Ellerbroek L, Johne R (2012) PCR inhibitors - occurrence, properties and removal. J Appl Microbiol 113: 1014-1026. [Crossref]
- Scipioni A, Mauroy A, Ziant D, Saegerman C, Thiry E (2008) A SYBR Green RT-PCR assay in single tube to detect human and bovine noroviruses and control for inhibition. Virol J 5: 94. [Crossref]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, et al. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901-906. [Crossref]
- Xu P, Guo M, Hay BA (2004) MicroRNAs and the regulation of cell death. Trends Genet 20: 617-624. [Crossref]
- Malumbres M (2013) miRNAs and cancer: an epigenetics view. Mol Aspects Med 34: 863-874. [Crossref]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, et al. (2007) Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129: 303-317. [Crossref]
- Phua YL, Chu JY, Marrone AK, Bodnar AJ, Sims-Lucas S, et al. (2015) Renal stromal miRNAs are required for normal nephrogenesis and glomerular mesangial survival. Physiol Rep 3. [Crossref]
- Maes OC, Chertkow HM, Wang E, Schipper HM (2009) MicroRNA: Implications for Alzheimer Disease and other Human CNS Disorders. Curr Genomics 10: 154-168. [Crossref]
- Li J, Li J, Liu X, Qin S, Guan Y, et al. (2013) MicroRNA expression profile and functional analysis reveal that miR-382 is a critical novel gene of alcohol addiction. EMBO Mol Med 5: 1402-1414. [Crossref]
- Romao JM, Jin W, Dodson MV, Hausman GJ, Moore SS, et al. (2011) MicroRNA regulation in mammalian adipogenesis. Exp Biol Med (Maywood) 236: 997-1004. [Crossref]
- Mencía A, Modamio-Høybjør S, Redshaw N, Morín M, et al (2009) Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet 41: 609-613. [Crossref]
- Hughes AE, Bradley DT, Campbell M, Lechner J, Dash DP, et al. (2011) Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am J Hum Genet 89: 628-633. [Crossref]
- de Pontual L, Yao E, Callier P, Faivre L, Drouin V, et al. (2011) Germline deletion of the miR-17∼92 cluster causes skeletal and growth defects in humans. Nat Genet 43: 1026-1030. [Crossref]
- Tuddenham L, Jung JS, Chane-Woon-Ming B, Dölken L, Pfeffer S (2012) Small RNA deep sequencing identifies microRNAs and other small noncoding RNAs from human herpesvirus 6B. J Virol 86: 1638-1649. [Crossref]
- Lu M, Zhang Q, Deng M, Miao J, Guo Y, et al. (2008) An analysis of human microRNA and disease associations. PLoS One 3: e3420. [Crossref]
- Gregory RI, Shiekhattar R (2005) MicroRNA biogenesis and cancer. Cancer Res 65: 3509-3512. [Crossref]
- Cummins JM, Velculescu VE (2006) Implications of micro-RNA profiling for cancer diagnosis. Oncogene 25: 6220-6227. [Crossref]
- Calin GA, Ferracin M, Cimmino A, Dileva G, Shimizu M, et al, (2005) A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Eng J Med 353: 1793-1801. [Crossref]
- Chen CZ (2005) MicroRNAs as oncogenes and tumor suppressors. N Engl J Med 353: 1768-1771. [Crossref]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, et al. (2004) Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101: 2999-3004. [Crossref]
- Schepeler T, Reinert JT, Ostenfeld MS, Christensen LL, Silahtaroglu AN, et al. (2008) Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res 68: 6416-6424. [Crossref]
- Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, et al (2008) MicroRNA expression profile associated with progression and therapeutic outcome in colon adenocarcinoma. J Am Med Assoc 299: 425-436. [Crossref]
- Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6: 857-866. [Crossref]
- Ansevin AT, Vizard DL, Brown BW and McConathy J (1976) High-resolution thermal denaturation of DNA. I. Theoretical & practical considerations for the resolution of thermal subtransitions. Biopolymers 1: 153-174. [Crossref]
- Ririe KM, Rasmussen RP, Wittwer CT (1997) Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 245: 154-160. [Crossref]
- Ansevin AT, Vizard DL, Brown BW, McConathy J (1976) High-resolution thermal denaturation of DNA. I. Theoretical and practical considerations for the resolution of thermal subtransitions. Biopolymers 15: 153-174. [Crossref]
- Freier SM, Kierzek R, Jaeger JA, Sugimoto N, Caruthers MH, et al. (1986) Improved free-energy parameters for predictions of RNA duplex stability. Proc Natl Acad Sci U S A 83: 9373-9377. [Crossref]
- Wittwer CT (2009) High-resolution DNA melting analysis: advancements and limitations. Hum Mutat 30: 857-859. [Crossref]
- Lay MJ, Wittwer CT (1997) Real-time fluorescence genotyping of factor V Leiden during rapid-cycle PCR. Clin Chem 43: 2262-2267. [Crossref]
- Wienken CJ, Baaske P, Duhr S, Braun D (2011) Thermophoretic melting curves quantify the conformation and stability of RNA and DNA. Nucleic Acids Res 39: e52. [Crossref]
- Dwight Z, Palais R, Wittwer CT (2011) µMELT: prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application. Bioinformatics 27: 1019-1020. [Crossref]
- Gudnason H, Dufva M, Bang DD, Wolff A (2007) Comparison of multiple DNA dyes for real-time PCR: effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res 35: e127. [Crossref]
- Oscorbin IP, Belousova EA, Zakabunin AI, Boyarskikh UA, Filipenko ML (2016) Comparison of fluorescent intercalating dyes for quantitative loop-mediated isothermal amplification. BioTechniques 61: 20-25. [Crossref]
- Horáková H, Polakovičová I, Shaik GM, Eitler J, Bugajev V, et al. (2011) 1,2-propanediol-trehalose mixture as a potent quantitative real-time PCR enhancer. BMC Biotechnol 11: 41. [Crossref]
- Dwight ZL, Palais R, Wittwer CT (2012) uAnalyze: web-based high-resolution DNA melting analysis with comparison to thermodynamic predictions. IEEE/ACM Trans Comput Biol Bioinform 9: 1805-1811. [Crossref]
- Varga A, James D (2006) Real-time RT-PCR and SYBR Green I melting curve analysis for the identification of Plum pox virus strains C, EA, and W: effect of amplicon size, melt rate, and dye translocation. J Virol Methods 132: 146-153. [Crossref]
- Mendes RE, Kiyota KA, Monteiro J, Castanheira M (2007) Rapid detection and identification of metallo-ß-Lactamase-encoding genes by multiplex real-time PCR assay and melt curve analysis. J Clin Microbiol 45: 544-547. [Crossref]
- Guion CE, Ochoa TJ, Walker CM, Barletta F, Cleary TG (2008) Detection of diarrheagenic Escherichia coli by use of melting-curve analysis and real-time multiplex PCR. J Clin Microbiol 46: 1752-1757. [Crossref]
- Winder L, Phillips C, Richards N, Ochoa-Corona F, Hardwick S, et al. (2011) Evaluation of DNA melting analysis as a tool for species identification. Meth Ecology Evol 2: 312-320.
- Von Keyserling H, Bergmann T, Wiesel M, Kaufmann AM (2011) The use of melting curves as a novel approach for validation of real-time PCR instruments. Biotechniques 51: 179-184. [Crossref]
- Bohling SD, Wittwer CT, King TC, Elenitoba-Johnson KS (1999) Fluorescence melting curve analysis for the detection of the bcl-1/JH translocation in mantle cell lymphoma. Lab Invest 79: 337-345. [Crossref]
- Downey N (2014) Interpreting melt curves: An indicator, not a diagnosis.https://www.idtdna.com/pages/decoded/decoded-articles/pcr-qpcr/decoded/2014/01/20/interpreting-melt-curves-an-indicator-not-a-diagnosis.
- Farrar JS, Reed GH, Wittwer CT (2014) High-resolution melting curve analysis for molecular diagnosis. In Molecular Diagnostics, 2nd Ed, pp. 229-245, Elsevier Ltd.
- Krypuy M, Ahmed AA, Etemadmoghadam D, Hyland SJ, Australian Ovarian Cancer Study Group, et al. (2007) High resolution melting for mutation scanning of TP53 exons 5-8. BMC Cancer 7: 168. [Crossref]
- Pasay C, Arlian L, Morgan M, Vyszenski-Moher D, Rose A, et al. (2008) High-resolution melt analysis for the detection of a mutation associated with permethrin resistance in a population of scabies mites. Med Vet Entomol 22: 82-88. [Crossref]
- Montgomery JL, Sanford LN, Wittwer CT (2010) High-resolution DNA melting analysis in clinical research and diagnostics. Expert Rev Mol Diagn 10: 219-240. [Crossref]
- Ahmed FE, Vos P, iJames S, Lysle DT, Allison RR, et al. (2007) Transcriptomic molecular markers for screening human colon cancer in stool & tissue. Cancer Genom Proteom 4: 1-20. [Crossref]
- Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15-20. [Crossref]
- Peterson SM, Thompson JA, Ufkin ML, Sathyanarayana P, Liaw L, et al. (2014) Common features of microRNA target prediction tools. Front Genet 5: 23. [Crossref]