Cancer is one of the major health problems with a high mortality rate. Apart from the genetic changes in cancer cells, epigenetic and environmental factors play an important role towards the progression of tumor. The new cancer cases worldwide are estimated to increase to 19.3 million per year by 2025, due to the changing lifestyle and increase in longevity. Social and economic changes have contributed to rising risks of cancers associated with dietary and hormonal factors. Although traditional therapies have been effective in cancer treatment, they often have adverse side effects due to their non specific action on both normal and tumor cells. Therefore, targeted treatment strategies using small molecule inhibitors are being extensively studied. These compounds are usually ≤500Da size and are often administered orally. Their small size also allows them to translocate through the plasma membrane and interact with the cytoplasmic domain of cell-surface receptors and intracellular signaling molecules. In principle, small molecule compounds can be developed to target any portion of a molecule, regardless of the target’s cellular location. In this review, we provide a summary of the small molecule inhibitors (SMIs) used in the cancer therapy, and elaborate on the recent advances in cancer therapies with emphasis on SMIs that block the growth of cancer cells.
cancer treatment; small inhibitors
The traditional means of cancer management are chemotherapy, radiation therapy and surgery. Chemotherapy that involves the use of a single drug at a time (single-agent chemotherapy) or several drugs (combination therapy) usually target killing of rapidly dividing cancer cells. Unfortunately, the chemotherapy also concomitantly targets normal cells such as cells in hair follicles, bone marrow and the digestive tract, thus resulting in adverse side effects for example, alopecia, anemia, fatigue, diarrhea, constipation and also include nausea and vomiting [1-6]. On the other hand, surgery that involves physically removing the macroscopic cancer along with some surrounding healthy tissue is usually done as a primary treatment to prevent cancer spread or to relieve the side effects of cancer associated inflammation. However, the surgery-related bleeding, damage to nearby tissues and drug reactions remain common, undesirable side effects of surgery. Moreover, current cancer treatment and management options often combine chemotherapy and radiation therapy. The radiation therapy involves the use of high energy waves to destroy the cancer cells, and similar to chemotherapy, can also cause damage to the epithelial surfaces, swelling of soft tissues and other intestinal discomforts. To overcome the drawbacks of traditional cancer therapies, the approach has been to search for specific molecular targets for selective elimination of cancer cells. Such targeted therapy would conceptually be more specific than the traditional non-targeted therapy. The two main approaches of specific molecular targeting available for use in clinical practice are small molecule agents and monoclonal antibodies (mAbs) [7]. When compared to mAbs which are usually large molecular weight proteins of around 150kDa, small molecule cancer drugs are much smaller in size (≤500Da) and thus can translocate through plasma membranes. On the contrary, the mAbs can only act on molecules that are expressed on the cell surface [8]. The small molecule inhibitors are also comparatively cost effective and are amenable to oral administration while the mAbs are mostly administered intravenously [9].
Cancer is a “miscommunication” disease, initiation and further progression of cancer relies on over activation of various extrinsic and intracellular signalling pathways. Small molecule cancer drugs, because of their small size, have been successfully used to target the extracellular, cell surface ligand-binding receptors as well as the intracellular proteins, including anti-apoptotic proteins that play a key role in transducing downstream signalling for cell growth and metastasis promotion.Research on molecularly targeted cancer drug discovery over the last few decades has resulted in a number of small molecule drugs being successfully introduced in the clinic for cancer treatment. Most of these drugs inhibit critical cancer targets such as serine/threonine/tyrosine kinases, matrix metalloproteinases (MMPs), heat shock proteins (HSPs), proteosome and other proteins playing a role in signal transduction pathways. Although a number of SMIs that target a variety of cell signaling molecules have been developed and currently being utilized in the clinic, the emergence of drug-resistant variants of cancers remain a significant and formidable problem that necessitates identification of additional, novel signaling molecules and exploitation of this knowledge for the development of additional strategies to effectively treat resistant cancers in the clinic.
Small molecule kinase inhibitors
Receptor tyrosine kinases (RTKs) are often preferred as key targets for anti-cancer drugs as aberrant activation of these usually result in downstream signalling that involves activation of pivotal cytoplasmic kinases which are usually serine/threonine kinases (STKs) [10]. Small molecule cancer drugs targeting both the extracellular RTKs and cytoplasmic STKs are thus being extensively studied.Deregulated activation of RTKs results in increased cell growth and survival, and often contributes to progression of cancer. For instance, HER2 (ErbB2), a constitutively active member of the epidermal growth factor receptor (EGFR) family of RTKs, is amplified and thus over expressed in a subset of breast cancers. On the other hand, over-expression of EGFR or its mutant version is often encountered in aggressive, non-small cell lung cancers (NSCLC). Overexpression and activation of vascular endothelial growth factor receptor (VEGFR) often contributes to cancer-associated angiogenesis and development. In most cases of cancer, overexpression or activation of RTKs results in constitutive gene expression to promote cell growth and survival that leads to progression of cancer. Receptor tyrosine kinases transmit the extracellular signals to the cytoplasm and subsequently to the nucleus leading to activation of an array of signalling pathways while non receptor tyrosine kinases that are found in the cytosol and the nucleus are activated by intracellular signals. Autophosphorylation and subsequent activation of tyrosine kinases by somatic mutation or binding of growth factor induced dimerization of RTKs can promote angiogenesis, constitutive expression of anti-apoptotic factors that collectively drive uncontrolled tumor cell proliferation leading to cancer metastasis and spread. The activated tyrosine kinases stimulate various downstream signalling cascades including the phospho-inositol 3’-kinase/Akt pathway, the Ras/Raf/mitogen-activated protein kinase pathway, the signal transducer and activator of transcription 3 (STAT 3) and the PLC-γ-CaMK pathway.
Several growth factor receptors with intrinsic tyrosine kinase activity have been implicated in the development and progression of neoplastic diseases. EGFRs (EGFR/HER1/ErbB1, EGFR2/HER2/ErbB2, EGFR3/HER3/ErbB3), VEGFR, platelet-derived growth factor receptor (PDGFR), hepatocyte growth factor receptor (HGFR), RET, insulin-like growth factor receptor (IGFR), and fibroblast growth factor receptor (FGFR) are classes of receptor tyrosine kinases that are usually constitutively active in most cancers and inhibition of these kinases using small molecule inhibitors has been found to sensitize the tumor cells to apoptosis [11]. Small molecule inhibitors of IGF-1R can be used to block IGF-1R function. It has been shown by preclinical studies that it is possible to reverse the neoplastic phenotype by down regulation of IGF-IR [12] . Various strategies have been pursued to inhibit tumorigenic effects of growth factors. Constitutive expression of VEGF, resulting in enhanced production of angiogenic signals is a requisite for tumor cells to achieve rapid growth. Angiogenesis inhibitors have been shown to have significant potential in cancer treatment, most notably prostate cancer [13]. Similarly, the epidermal growth factor receptor (EGFR) is also a target for therapeutic intervention and it is possible to inhibit EGFR downstream signaling by either blocking the intracellular kinase activity with small-molecule tyrosine kinase inhibitors (TKIs) or by using antibodies directed against the extracellular domain of the receptor thus preventing binding of the ligand [14]. The first anti-receptor drug to be developed was against the HER2,which is a type 1 transmembrane RTK found to be overexpressed in many cancers [15,16]. Mutations culminating in constitutive activation of EGFR have been associated with many human tumors including tumors of the breast, ovaries, kidneys, laryngeal, esophageal, gastric, pancreatic, colon, renal cell, bladder, and non small cell lung cancer [17]. Hence, many EGFR-TKIs have been developed and are being successfully used in clinic while others such as canertinib and vatalanib are being tested in clinical trials.In spite of various haematological side effects; TKIs have been found to be well tolerated. Although drugs have been discovered that target cell surface receptors and attenuate/inhibit its activation and intracellular signaling, the cancer cells often develop resistance by switching to alternate signaling pathways and thus necessitate identification of strategies to overcome resistance.
Non receptor tyrosine kinases including Src, janus-activated kinases (JAKs), c-Abl and focal adhesion kinase (FAK) have also been identified as potential anticancer targets as they transmit the intracellular signals and several small molecule drugs targeting these are being developed. BCR-Abl, an oncogene responsible for causing chronic myelogenous leukemia (CML) was identified as a key target for drug intervention. Imatinib, approved in 2001, was the first BCR-Abl inhibitor used in treatment of CML. Nilitonib, bosutinib, ponatinib and dasatinib are the second generation BCR-Abl inhibitors that were subsequently developed and are currently used in clinic [18].
Serine/threonine kinases are a class of protein kinases which have not received as much attention as the tyrosine kinases, as potential anti-cancer targets. They have been shown to activate cell cycle regulators, cytoplasmic and nuclear effectors and other significant transcription factors [19]. Thus, aberrant expression of the serine threonine kinases may be implicated in cancer progression. For instance, the serine threonine kinase, Aurora-2 which is a key cell cycle regulator was shown to be overexpressed in many human tumors, most notably in pancreatic cancer [20]. Akt, another serine-threonine kinase, controls cellular survival and various metabolic processes. It has also been shown to confer resistance to tumor cells against chemotherapy by inducing survival signals [21]. Blocking Akt signalling could therefore inhibit proliferation of tumor cells and also sensitize them to apoptosis. Akt/protein kinase B signaling inhibitor-2 (API-2), a small molecule inhibitor has been shown to inhibit Akt kinase thus inducing apoptosis in human cancer cells [22]. Perifosine is a novel oral inhibitor of Akt pathway that has entered into phase 3 of clinical trial and found to inhibit Akt activation in hepatoma cells [23]. PI3K/Akt/mTOR pathway plays a pivotal role in cancer, and drugs targeting various components of the pathway such as PI3K, Akt and mTORC1/2 have been developed. Everolimus, the mTORC1 inhibitor is being used as a therapeutic in various cancers. Buparlisib (BKM120), the pan PI3K inhibitor; BEZ235, the dual pan-PI3K/mTORC1/2 inhibitor and BYL719 are novel compounds that are in clinical development [24]. Phosphorylation of Raf serine threonine kinase as a result of ligand induced activation of receptor tyrosine kinases plays a pivotal role in relaying the intracellular signal to the nucleus via the Ras-Raf-MEK-ERK pathway. It is thus possible to block the aberrant signalling by inhibiting Raf using small molecule inhibitors such as sorafenib. Vemurafenib, another B-Raf/MEK/ERK pathway inhibitor used in the treatment of late stage melanoma received FDA approval in 2011, making it the first drug developed using fragment-based lead discovery to gain regulatory approval [25]. Protein kinase C inhibitors such as Midostaurin (PKC412) and Enzastaurin (LY317615) are in various phases of clinical trials [26]. Other serine/threonine kinases including phosphorylase kinase, CaM kinases and protein kinase A have also been identified as potential cancer targets and new small molecule inhibitors targeting these can be expected to be developed in near future.
Small molecule proteosome inhibitors
It is intriguing to note that pathways that help to maintain cellular homeostasis in normal cells are those that favour cancer cell survival by up regulating cell proliferation and inhibiting apoptosis of tumor cells. The ubiquitin proteosome pathway (UPP) is one such pathway that involves degradation of various cell cycle regulators and anti-apoptotic proteins, and has been identified as one of the crucial targets of cancer therapy. UPP is involved in proteolysis of various cyclins and cyclin dependent kinases thus assisting in transition of cell cycle from one phase to other [27]. Further, the UPP has also been shown to control the expression of cyclin-dependent kinase (CDK) inhibitor p27, an important cell cycle regulator, low levels of which has been associated with various tumors including lung, colon, breast, brain, ovarian and lymphoma [28]. Resistance of tumor cells to apoptosis which also confers resistance to chemotherapy is another major factor contributing to tumor cell survival and progression. The UPP inhibits apoptosis of tumor cells by regulating expression of various proteins involved in apoptosis such as inhibitor of apoptosis (IAP) proteins and some caspases [29]. Hence it was assumed that preventing degradation of these cell cycle regulators and apoptotic regulators using proteosome inhibitors will culminate in cell cycle arrest and also sensitize the cells to apoptosis. Proteosome inhibitors have been studied to effectively induce apoptosis of tumor cells by increasing the expression of pro-apoptotic factors and reducing expression of anti-apoptotic IAP proteins [30]. In this regard, a small molecule proteasome inhibitor,bortezomib (Velcade) has been developed and being used in clinic for treatment of renal carcinoma, mantle cell lymphoma, and multiple myeloma. Bortezomib was shown to promote apoptosis in cancer cells in a manner dependent on the tumor suppressor p53 gene [31,32].
UPP plays a major role in oncogenic NF-kB activation as it regulates degradation of Ik-B, the inhibitory partner protein of NF-kB [33]. NF-kB is a transcription factor that has been found to play a major role in development of cancer as it is involved in the control of cell proliferation, inflammation and inhibition of apoptosis [34,35]. NF-kB also participates in processes of angiogenesis and metastasis, which are essential for tumor progression [34]. Inhibition of NF-kB is thus an effective strategy to induce tumor cell apoptosis. In the cytoplasm, I-B remains bound to NF-B thus preventing the translocation of NF-B into the nucleus and further transcription of growth and metastasis promoting genes. Preventing the degradation of I-B by the UPP remains a major strategy to keep NF-B in an inactive state. Carfilzomib, another proteosome inhibitor has been approved by the FDA for treatment of multiple myeloma in patients who have received prior treatment with bortezomib [36]. The second generation proteosme inhibitors, oprozomib (ONX 0912), ixazomib (MLN9708), marizomib (NPI-0052), and delanzomib (CEP-18770) are in various phases of clinical trial. The advantage of these inhibitors over bortezomib is that they are orally administered [37]. During the preclinical and clinical studies, the efficacy of UPP targeting drugs was shown to be reduced by certain natural products such as green tea polyphenols [38]. The use of UPP-targeting drugs was also associated with certain adverse effects such as peripheral neuropathy and thrombocytopenia. Disturbingly, development of drug resistance was also a major limitation which was observed even in patients receiving bortezomib monotherapy for the first time [39].
Small molecule MMPs and HSPs inhibitors
Matrix metalloproteinases (MMPs) are a group of endopeptidases that degrade the extracellular matrix (ECM), thus paving the way for cell migration and invasion [40]. More than 20 MMPs have been identified in humans, the activities of which are regulated in vivo by the tissue inducers of metalloproteinases (TIMPs). The gelatinases MMP-2 and MMP-9 have been found to target many signaling pathways therefore aggravating cancer cell migration and invasion [41]. The use of MMP inhibitors as therapeutics in cancer has been explored but was shown to have limited clinical success [42]. Batimastat (BB-94) was the first MMP inhibitor, the effects of which were explored in cancer patients. Though proved to be a potent inhibitor, it was found to be non specific and was not orally bio-available [43]. BMS-275291, that was found to inhibit MMP-1,-2,-3,-8,-9,-13 and MMP-14, failed in phase 3 trial when used in combination with paclitaxel and carboplatin for treatment of advanced non small cell lung cancer [44]. Tanomastat (BAY12-9566), Marimastat (BB-2516), Neovastat (AE-941), Prinomastat (AG-3340) are MMPIs which progressed till phase III of clinical trial [45]. The effects of specific inhibitory antibodies to be developed into an effective MMP inhibitor were also studied. DX-2400, a selective human antibody inhibitor of MMP-14 was shown to inhibit tumor growth and metastasis in vivo [46]. The major drawback of these MMP inhibitors was that they non-specifically bind to broad spectrum of MMPs, thus also inhibit a few MMPs which have been identified to possess cancer-inhibiting function [47]. MMPIs can be developed and used as potential anticancer agents by selectively targeting cancer associated MMPs, thus minimizing the adverse side effects.
Heat shock proteins (HSPs) are chaperones that help with proper folding and transport of proteins across the cell membrane. HSPs are implicated with progression and aggression of cancer as they help in proper folding and transport of oncoproteins which play important role in many signaling pathways. Expression of HSP 27, 70 and 90 has been found to be abnormally high in various cancers which in turn is associated with resistance to chemotherapy and inhibition of apoptosis [48]. Among these, HSP90 has been identified as a particularly potential cancer target as various mutant forms of oncogenic signaling proteins that are elevated in many cancers have been found to be highly dependent on HSP90 for their stability and function [49]. For instance, oncogeneic proteins HER2 and c-Src which are considered to play important role in progression of breast cancer have been found to be over dependent on HSP90 [50]. Geldanamycin, a benzoquinone ansamycin antibiotic inhibits the function of Hsp90 and thus impact expression and functions of HSP90 client oncogenes. Despite its potent antitumor potential, geldanamycin caused significant hepatotoxicity. Accordingly, geldanamycin analogues such as 17-N-allylamino-17-demethoxygeldanamycin (17-AAG; Tanespimycin) were developed, tested in the phase 2 trials, and its development as anti-melanoma agent was unfortunately halted in 2010. Ganetespib, the novel small molecule inhibitor of HSP90 has been shown to exhibit anti-tumor effects against various cancers and is under investigation as a cancer therapeutic [51]. MPC-3100, KW-2478, AT13387, NVP-AUY922 are other HSP90 inhibitors that are in various phases of clinical trial [52]. Inspite of being identified as a potential drug target, not a single small molecule inhibitor of HSP90 has thus far been approved for clinical use. The main drawbacks of first generation HSP90 inhibitors were that they were highly toxic, less potent and they did not exhibit significant clinical activities. Further studies elucidating the molecular structure and function of HSP90 along with a proper understanding of molecular mechanisms of its association with oncogeneic proteins will likely pave the way for development of improved, novel inhibitors of HSP90 with increased therapeutic potential.
Small molecule inhibitors targeting the apoptosis
Small molecule drugs that potentially restore the wild type tumor suppressor functions to the mutated p53 have been developed. CP-31398, Phikan083, the tenovins, MI-219 and the nutlins are some of the small molecules that target the mutated p53 and induce apoptosis of cancer cells [53-57]. Another class of small molecule drugs are the caspase activators that induce apoptosis by activating procaspase 3 and thus resulting in downstream activation of other caspases [58]. Bcl-2 family of proteins possesses a hydrophobic groove to which other proteins bind so as to activate the anti-apoptotic pathway [59]. Gossypol,apogossypolone,TW-37,ABT-737,ABT-263 (navitoclax) andGX-015-070(Obatoclax) are small molecule inhibitors of Bcl-2 family proteins that have been shown to induce apoptosis of various cancer cells by blocking their anti apoptotic effects [60]. Gossypol, apogossypolon and TW-37 were specifically shown to effectively inhibit growth and invasion of pancreatic cancer cells [61-63].
Small molecule mimetics of regulatory proteins involved in inhibiting tumor cell growth can also be developed as potential cancer treatments. In this context, CARP-1/CCAR1 was originally identified as a novel regulator of apoptosis when cells were treated with chemotherapeutics Adriamycin, etoposide, Gefitinib, and a novel, apoptosis-promoting retinoid. Subsequently, CARP-1 was also identified as a zyxin-binding partner that functions in transducing radiation-induced apoptosis [64]. CARP-1 is a well-known, biphasic regulator of cell growth and survival signaling by EGFR receptors and steroid-thyroid nuclear hormone receptor family and β-catenin. Although, several advanced and computational techniques are being developed and utilized to identify novel small molecule inhibitors of various signaling proteins [65,66], a high-through-put screening strategy was utilized to identify small molecule inhibitors of CARP-1 interaction with cell cycle regulatory anaphase promoting complex/cyclosome (APC/C). Screening of a chemical library led to discovery of multiple small molecule inhibitors of CARP-1 binding with APC/C subunit APC-2. These compounds, termed CARP-1 Functional Mimetics (CFMs) bind with CARP-1 and prevent its interaction with APC-2, and cause cell cycle arrest and apoptosis. CFMs promote apoptosis and inhibit growth of a number of cancer cell types including the breast cancer cells that are resistant to doxorubicin or tamoxifen, without inhibiting growth of non-tumorgenic human breast epithelial MCF-10A cells, underscoring their potential for therapeutic application. Apoptosis by CFMs involves elevated level of CARP-1, down-regulation of G2M cyclin B1, and activation of various stress-activated protein kinases (SAPKs). CFMs have been shown to effectively inhibit growth of breast, colon, prostate, medulloblastoma and neuroblastoma cancer cells by activation of caspases 8, 9 and 3 and apoptosis [67,68] while simultaneously attenuating levels of cyclin B1 [69]. CFMs also inhibit growth of malignant pleural mesothelioma (MPM) cells and could be used to treat the lethal form of asbestos related malignancy [70]. These studies have shown the efficacy of CFMs to be developed into potential anti-cancer drugs (Figure 1).
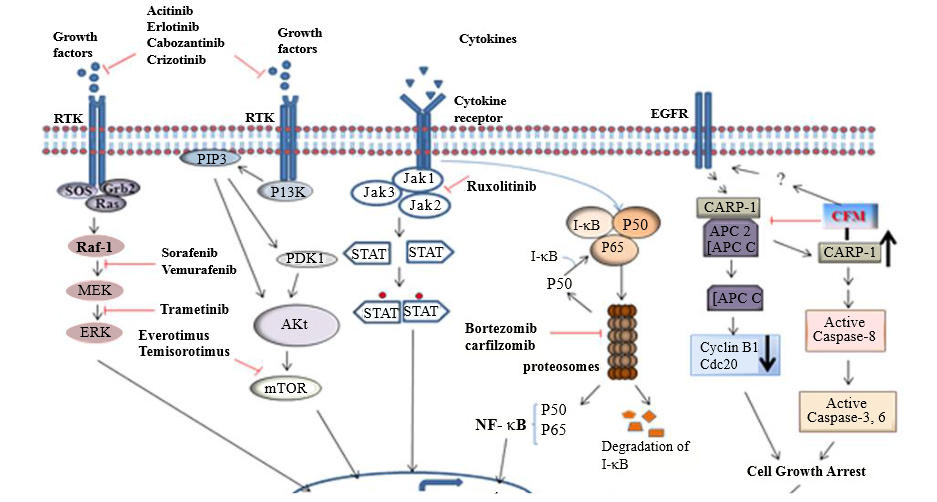
Figure 1: Summarized known small molecule inhibitors as targeted therapy. Schematic representation of pathways that are usually aberrantly activated in cancer, along with the small molecule inhibitors of critical cancer targets
Many cancer therapeutic drugs have been approved for use and several more are being studied in clinical trials. There are various stages, after which a drug is commercially available in the market. After being discovered and developed in the laboratory, the drugs are subjected to preclinical research after which they are tested in humans to evaluate their efficacy and safety. The US FDA review team then makes a thorough study of all the data before approving the drug for marketing. There are regulations for approval of drugs which are different in different countries. Majority of small molecule inhibitors currently in use were initially approved in US, later in European Union and finally in India. Like the US FDA, the main regulatory body in India is the Central Drugs Standard Control Organization(CDSCO).The CDSCO is controlled by The Drug Controller General of India (DCGI) [71] (Table 1).
Table 1: List of small molecule inhibitors for cancer therapy
| Molecular Target |
Small Molecule Drug |
Specific Target |
Cancer Targeted |
Clinical Status |
Reference |
| 1.Tyrosine & serine/threonine Kinases |
Imatinib
(Gleevec) |
Bcr-Abl |
Philadelphia chromosome-positive chronic myelogenous leukemia |
FDA approved in 2001 |
[69] |
Gefitinib
(Iressa) |
EGFR |
Non small cell lung cancer |
FDA approved in 2003 |
[70] |
Erlotinib
(Tarceva) |
EGFR |
Non small cell lung cancer, pancreatic cancer |
FDA approved
in 2005 |
[71] |
Sunitinib
(sutent) |
VEGFR2,RET
PDGFR, FLT-3, KIT, CSF-1 |
Renal cell carcinoma |
FDA approved in 2006 |
[72] |
Lapatinib
(Tykerb) |
EGFR, HER2/neu |
Breast cancer |
FDA approved in 2007 |
[73] |
Nilotinib
(Tasigna) |
Bcr-Abl,
KIT,LCK |
Chronic
myelogenous leukemia |
FDA approved in 2007 |
[74] |
Sorafenib
(Nexavar) |
B-Raf, VEGFR2
EGFR,PDGFR |
hapatocellular carcinoma |
FDA approved in 2007 |
[75] |
| Temsirolimus(CCI-779) |
mTOR |
Renal cell carcinoma |
FDA approved in 2007 |
[76] |
Everolimus
(afinitor) |
mTOR |
Renal cell carcinoma |
FDA approved in 2009 |
[77] |
| Pazopanib (Votrient) |
c-KIT, FGFR, PDGFR and VEGFR |
Renal cell carcinoma, soft tissue sarcoma |
FDA approved in 2009 |
[78] |
Crizotinib
(Xalkori) |
HGFR |
Non small cell Lung Cancer |
FDA approved in 2011 |
[79] |
Ruxolitinib
(jafaki) |
Jak1,Jak2 |
Primary Myelofibrosis |
FDA approved in 2011 |
[80] |
| Vandetenib (Caprelsa) |
VEGFR, EGFR,RET |
Medullary thyroid cancer |
FDA approved in 2011 |
[81] |
Axitinib
(Inlyta) |
VEGFR1-3, cKIT, PDGFR |
Renal cell carcinoma |
FDA approved in 2012 |
[82] |
Bosutinib
(Bosulif) |
Src, Bcr-Abl |
Philadelphia chromosome-positive chronic myelogenous leukemia |
FDA approved in 2012 |
[83] |
Cabozantinib
(Cometriq) |
c.Met, VEGFR2 |
Medullary thyroid cancer |
FDA approved in 2012 |
[84] |
Ponatinib
(Iclusig) |
Bcr-Abl |
Chronic myeloid leukemia, acute lymphoblastic leukemia |
FDA approved in 2012 |
[85] |
| Regorafenib (Stivagra) |
VEGFR1-3, c-Kit, TIE-2, PDGFR-β, FGFR-1, RET, Raf-1, B-RAF |
Metastatic colorectal cancer |
FDA approved in 2012 |
[86] |
| Ibrutinib (Imbruvica) |
Bruton’s tyrosine Kinase |
Mantle cell lymphoma |
FDA approved in 2013 |
[87] |
| Trametinib (Mekinist) |
MEK |
B-RAF 600E/K-mutant metastatic melanoma |
FDA approved in 2013 |
[88] |
| |
Perifosine
|
Akt |
Colorectal Cancer, Multiple Myeloma, Colon Cancer |
Approved as Orphan drug for multiple myeloma and neuroblastoma.
Phase II of clinical trial for metastatic colon cancer. |
[89] |
| 2. Proteosomes |
Bortezomib
(Velcade) |
26S proteosome |
Multiple myeloma |
FDA approved in 2003 |
[90] |
| Carfilzomib (Kyprolis) |
20S proteosome |
Multiple myeloma |
FDA approved in 2012 |
[91] |
| Marizomib (NPI-0052) |
20S proteosome |
Multiple myeloma |
Granted orphan drug status in 2014 |
[92] |
3.MMPs and
HSPs
|
Batimastat (BB-94) |
Broad spectrum MMPs |
Various tumors |
Yet to be approved |
[40] |
| Neovastat (AE-941) |
MMPs 2,9,12; VEGFR-2 |
Renal cell carcinoma, non small cell lymphoma |
Phase III of clinical trial |
[42] |
| Prinomastat (AG-3340) |
MMPs 2, 3, 9, 13, and 14 |
renal cell carcinoma |
Phase III of clinical trial completed |
[93] |
Rebimastat
(BMS-275291) |
MMPs 1, 2, 8, 9, and 14 |
advanced non small cell lung cancer |
Phase III of clinical trial |
[41] |
| Ganetespib |
HSP 90 |
Multiple cancers |
Phase III of clinical trial |
[48] |
| NVP-AUY922 |
HSP 90 |
Various tumors |
Phase II of clinical trial |
[94-96] |
| |
Marimastat (BB-2516) |
Broad spectrum MMPs |
- |
development terminated due to poor performance in clinical trial |
[97] |
| 4.Apoptosis |
Obatoclax (GX15-070) |
Bcl-2 family of proteins |
Leukemia, lymphoma, myelofibrosis |
Phase II of clinical trial |
[98] |
| Navitoclax(ABT-263) |
Bcl-xL, Bcl-2 and Bcl-w |
lymphoma and chronic lymphocytic leukemia |
Phase II of clinical trial |
[99] |
More than 20 small molecule drugs have been approved for clinical use and are being successfully used in cancer treatment. Still, there are certain limitations that are to be considered and overcome while designing more active drugs so as to reduce the failure rates of the drugs at the clinical level. Certain small molecule inhibitors bind to multiple molecular targets including cell surface receptors and other intracellular proteins thus increasing the risk of toxicity [103]. Most of the small molecule drugs have a short life span, thus requiring daily dosing which is not the case with therapeutic mAbs that have a longer life span [104]. Alternate ways such as coupling of these drugs to other compounds such as nano-carriers so as to increase the specificity and life span (bio-availability) can be considered.
Adverse side effects following treatment with small molecule drugs have also been reported. For instance, treatment with imatinib can cause oedema, diarrhoea, vomiting, nausea and sometimes can lead to thrombocytopenia, anaemia and neutropenia [105]. Drugs that inhibit angiogenesis can affect the formation of new blood vessels throughout the body that can lead to dangerous side effects such as bleeding that becomes difficult to manage [106]. Anti-EGFR inhibitors can target the skin cells, which often express abundant EGFR thus inhibiting the normal growth of skin cells and making it harder for the skin to retain moisture, resulting in scaly and itchy skin [107]. Nilotinib, used in the treatment of chronic myelogenous leukemia carries a warning for possible heart complications [108]. Development of resistance to these drugs is another major drawback and cancer cells acquiring resistance to drugs such as crizotinib and vemurafenib have been reported [109,110]. BRAF inhibitors Vemurafenib and dabarafinib although have revolutionized the treatment of melanoma, approximately a fifth of the patients develop cutaneous squamous cell carcinoma (cSCC) during therapy. A recent report revealed that these BRAF inhibitors suppress apoptosis through their off-target inhibition of pro-apoptotic JNKs while concomitantly and paradoxically activating ERKs [111]. Newer techniques to overcome drug resistance such as encapsulating the drugs in nanoparticles can be explored [112].
Over the last few decades, the success of small molecule cancer drugs over conventional chemotherapy has been clearly demonstrated. The main focus of molecularly targeted therapy using small molecule inhibitors has been the pathways that are usually deregulated in cancer, thus inhibiting cancer cell survival and proliferation. Majority of the inhibitors that have been developed and currently in clinical use target the kinases, which include the receptor molecules as well as downstream regulators [113]. With the exception of UPP inhibitor Bortezomib, small molecule inhibitors of MMPs and those targeting apoptosis have albeit been extensively studied, but are yet to be approved for clinical use [42,114]. Identifying specific genes/proteins, and understanding the mechanism(s) underlying the progression of each cancer will help design novel strategies to further improve the efficacy of current drugs and possibly identification of novel agents. The advantages of using a combination of different agents that inhibit several pathways or use of small molecule inhibitors in combination with radiation therapy can also be explored. In this regard, combination therapies using small molecule drugs and mAbs have been exploited and are emerging to be a promising anti-cancer strategy [2,115].
The rate at which new drugs are discovered and developed is frustratingly slow, with an increasing failure rate of most drugs at the clinical level. It is therefore of utmost importance to address the limitations of these drugs so as to reduce the delay in approval of these drugs for clinical use. With more than 60% of cancer deaths occurring in low and middle income countries, newer approaches are urgently warranted to identify and develop cost effective drugs. Future studies on small molecule cancer drugs should also focus on alternative strategies so as to develop newer drugs targeting novel pathways while striving to improve the efficacy of currently marketed drugs.
The authors gratefully acknowledge support by the fellowship from B.S. Abdur Rahman University, Chennai, India (SJ) and the United States Department of Veterans Affairs Merit review grant (AKR).