The phosphatidylinositol-3-kinase-Akt (protein kinase B) - mechanistic target of rapamycin (PI3K-Akt-mTOR) pathway is often dysregulated in cancer, including hematological malignancies. Primary acute myeloid leukemia (AML) cell populations may include various subclones at the time of diagnosis. A relapse can occur due to regrowth of the originally dominating clone, a subclone detectable at the time of first diagnosis, or a new clone derived either from the original clone or from remaining preleukemic stem cells. Inhibition of mTOR signaling has in general modest growth-inhibitory effects in preclinical AML models and clinical trials. Therefore, combination of allosteric mTOR inhibitors with standard chemotherapy or targeted agents has a greater anti-leukemia efficacy. Dual mTORC1/2 inhibitors, and dual PI3K/mTOR inhibitors show greater activity in pre-clinical AML models. Understanding the role of mTOR signaling in leukemia stem cells is important because AML stem cells may become chemoresistant by displaying aberrant signaling molecules, modifying epigenetic mechanisms, and altering the components of the bone marrow microenvironment. The PI3K/Akt/mTOR signaling pathway is promising target in the treatment of hematological malignancies, including AML, especially by using of combinations of mTOR inhibitors with conventional cytotoxic agents.
acute myeloid leukemia, Akt, bone marrow microenvironment, mTOR, leukemia stem cell, PI3K, allosteric mTOR inhibitors, dual PI3K/mTOR inhibitors
The mammalian target of rapamycin (mTOR) is a serine/threonine phosphatidylinositol 3´-kinase-related protein kinase involved in the contol of cellular growth and proliferation, protein synthesis and breakdown, cellular aging and survival, membrane trafficking, metabolism and development. This mammalian protein kinase, which is also called FK506-binding protein (FKBP12), rapamycin-associated protein 1 (FRAP1), rapamycin and FKBP12 target (RAFT1), rapamycin target (RAPT1), and sirolimus effector protein (SEP), was identified and cloned in 1994 [1-3]. The phosphatidylinositol 3-kinase (PI3K)/Akt (protein kinase B) /mTOR signaling pathway promotes tumorigenesis by promotion of cell proliferation and survival and by inhibition of apoptosis [4,5]. Rapamycin (sirolimus), a natural antibiotic, is US Federal Drug Administration (FDA)-approved as an immunosuppressive agent in solid organ transplantation. Sirolimus has antitumor activity that led to the identification of mTOR as a potential target for chemotherapy. Sirolimus has been used in several phase II-III trials against a variety of cancers as the first mTOR inhibitor [6]. Poor aqueous solubility and variable bioavailability of sirolimus caused a development of second-generation of mTOR inhibitors, including rapamycin analogues such as CCI-779 (temsirolimus), RAD001 (everolimus) and AP23573 (also known as ridaforolimus, deforolimus, and MK-8669) to overcome these difficulties [7]. Rapamycin derivatives are modified by the addition of an ester, ether or phosphonate group. Temsirolimus is a pro-drug of rapamycin administered intravenously. Temsirolimus was the first mTOR inhibitor to gain FDA approval in 2007 for patients with advanced or metastatic renal cell carcinoma [7-10].
Studies have shown that the PI3K/Akt/mTOR pathway is constitutively activated in 50-70% of acute myeloid leukemia (AML) cases, suggesting that this pathway could constitute a therapeutic target [11-41]. Constitutive activation of PI3K/Akt/mTOR pathway may be also involved in the pathophysiology of myelodysplastic syndromes, chronic myelogenous leukemia (CML), multiple myeloma and lymphoid leukemias and lymphomas [42-54]. Below, I discuss the PI3K/Akt/mTOR pathway and its role in AML. Clinical efforts relating to the use of mTOR inhibitors as single agents or in combination with PI3K, Akt and AMP-activated protein kinase (AMPK) inhibitors or with conventional cytotoxics and other targeted therapies in AML is also reviewed.
The mTOR gene maps to human chromosome 1p36.2. The mTOR protein contains multiple domains with highly conserved sequence and positions throughout evolution. The structure of mTOR potein is schematically shown in Figure 1. HEAT domains at the N-terminus of mTOR contain Huntington, elongation factor 3, the A subunit of protein phosphatase 2A (PP2A), and TOR motifs. C-terminus contains an FAT (the rapamycin-associated protein FRAP - ataxia telangiectasia mutated) domain [55]; an FRB (FKBP12-rapamycin binding) domain; a catalytic serine/threonine phosphatidylinositol 3´-kinase-related protein kinase (PIKK) domain; a probable autoinhibitory or repressor domain; and an FATC (FATcarboxy-terminal) domain. PIKK are involved in many critical regulatory cell cycle functions (cellular homeostasis, DNA damage, DNA repair, and DNA recombination). Rapamycin and its derivatives (CCI-779, AP23573 and RAD001) bind to FKBP12, an abundant intracellular protein [56]. This complex binds to FRB domain of mTOR and inhibits its kinase activity.
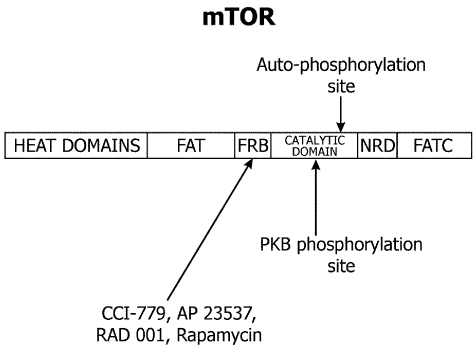
Figure 1: The structure of mammalian target of rapamycin (mTOR). The first 1200 amino acids consists of domains named for Huntington, elongation factor 3, the regulatory A subunit of protein phosphatase 2A, and Tor 1p (HEAT domains). The motif consists of stretches of cca 40 amino acids in at least 3 repeats and displays a consensus pattern of hydrophobic, proline, aspartic acid, and arginine residues. PI3K-related protein kinases possess a short segment at their extreme carboxyl terminus, termed the rapamycin – associated protein (FRAP) – ataxia telangiectasia mutated (ATM)-transformation/transcription domain-associated protein (TRRAP), carboxy-terminal homology domain (FATC domain) and a region of weaker homology between amino acids 1382-1982 in the human FRAP/mTOR (FAT domain). Because the FAT domain always is found in combination with the FATC region, it has been postulated that intramolecular interactions between FAT and FATC modulate kinase activity. The FK506 binding protein binding protein (FKBP12)/rapamycin –binding domain (FRB) lies immediately amino terminal to the kinase domain and downstream of the FAT domain. PKB: protein kinase B.
PI3Ks and their regulation
Insulin and other growth factors regulate mTOR activity. Growth factor-induced activation of mTOR is mediated by PI3K [57-59]. PI3Ks are divided into three classes based on their structure and sequence homology [60]. Class I PI3Ks are heterodimeric proteins, which have great significance for cancer. Therefore, class I PI3Ks attracted the major interest. The 110 kDa catalytic subunit consists of a catalytic lipid kinase domain, a Ras binding domain, a C2 phospholipid binding domain (protein kinase C homology domain), a helical PI kinase (PIK) domain and N-terminal domain, which forms a tight association with the 85 kDa regulatory subunit containing two Src-homology 2 domains [61]. Class I PI3Ks are activated by growth factor receptor tyrosine kinases or by G-protein-coupled receptors. Once activated, class I PI3Ks are recruited to the plasma membrane and bring the protein into close proximity with its substrate, the inositol phospholipid - phosphatidylinositol (4,5)-bisphosphate (PIP2) to produce the biologically active lipid, the secondary messenger phosphatidylinositol-3,4,5- trisphosphate (PIP3).
The deactivation of PI3K signaling is regulated by the tumor suppressor protein PTEN (phosphatase and tensin homologue deleted on chromosome 10), which specifically dephosphorylates PIP3 at the 3-position of the inositol ring to generate PIP2, thereby terminating the lipid signaling [62,63].
Akt-dependent mTOR regulation
PIP3 binds to the pleckstrin homology (PH) domain of the serine/threonine kinase Akt (according to v-Akt – murine thymoma viral oncoprotein homolog), promoting its translocation to the cell membrane. Akt (protein kinase B) is then activated by sequential phosphorylation at T308 within the activation loop and at S473 within C-terminal hydrophobic motif of Akt. Phosphorylation at T308 is mediated by PDK-1 (phosphoinoside-dependent kinase 1, a protein serine/threonine kinase that has multiple substrates), which itself is activated by the binding of PIP3 to its pleckstrin homology domain and subsequent translocation to the cell membrane [64-67]. Many kinases are able to phosphorylate Akt at serine 473 in vitro. However, Sarbassov et al. [68] showed that the mammalian target of rapamycin complex 2 (mTORC2) is the kinase responsible for S473 phosphorylation in vivo.
The complex mTORC2 consists of mTOR, mammalian stress-activated protein kinase interacting protein 1 (mSIN1), rapamycin-insensitive companion of mTOR (rictor) and a protein associated with rictor (protor). mTORC2 is often referred to as the rapamycin-insensitive complex of mTOR. However, prolonged exposure to rapamycin in certain cell types decreases Akt phosphorylation at S473 and may impair mTORC2 assembly and hence Akt activation [69]. Suppression of mTORC2 assembly was also observed in leukemic cells treated with rapamycin derivatives both in vitro and in a clinical trial in vivo [70].
Akt (protein kinase B) has many substrates within the cell. Phosphorylation of two substrates, TSC2 (tuberous sclerosis complex 2) and PRAS40 (proline-rich Akt substrate 40 kDa), leads to activation of mTOR. Akt activates the multiprotein complex mTORC1 by direct phosphorylation of the tumor suppressor TSC2 at S939 and T1462 [71,72]. TSC2 (tuberin) forms a heterodimeric complex with TSC1 (hamartin) in vivo [73,74]. This complex binds and inhibits mTOR. Phosphorylation of TSC2 at S939 and T1462 inhibits GTPase-activating protein activity of this complex. TSC2 suppresses the activity of the Ras-related GTPase Rheb, which is a selective activator of mTORC1 signaling [75,76]. Bai et al. [77] identified the protein FKB38, a mitochondrial membrane protein and a member of the immunophilin family, FK506 (tacrolimus)-binding protein 38, as a direct binding partner for GTPase Rheb. FKB38 binds to mTOR within the complex mTORC1 and serves as effector of Rheb. Inhibition of TSC2 by Akt results in activation of mTORC1, which impairs inhibition of mTOR activity possibly through dissociation of the TSC1-TSC2 complex [78].
Raptor (regulatory-associated protein of mammalian target of rapamycin) is critical for the regulation of mTORC1 activity in response to insulin signaling and nutrient and energy sufficiency. mTOR phosphorylates raptor in vitro and in vivo [79]. The phosphorylation of raptor at S859 and S863 is stimulated by insulin and inhibited by rapamycin. The site-directed mutation of raptor at one phosphorylation site, S863, reduced mTORC1 activity both in vitro and in vivo [79]. Moreover, the S863 mutant prevented small GTP-binding protein Rheb from enhancing the phosphorylation of S6 kinase in cells. Therefore, mTOR-mediated raptor phosphorylation plays an important role on activation of mTORC1.
Another characterized mTOR-interacting protein from S. cerevisiae Lst8p-its mammalian ortholog is mLST8/Gβ L (G protein β-subunit-like protein)-a highly conserved 36-kDa protein that consists almost entirely of seven WD-40 repeats with high sequence similarity to those found in the β subunits of heterotrimeric G proteins [79,80]. Loewith et al. reported that Lst8p interacts with Tor1p and Tor2p and showed that transiently expressed recombinant mTOR and mLST8 can interact [80]. Kim et al. [81] independently identified the same interacting protein and adopted the Gβ L name based on a previous report [82].
Akt also activates mTORC1 by a TSC2-independent mechanism. Akt directly phosphorylates PRAS40, a protein associated with mTORC1, and decreases its inhibitory effect on mTORC1 [83,84]. Akt can directly phosphorylate mTOR at S2448 in response to insulin stimulation [85]. It seems unlikely that this phosphorylation of mTOR at S2448 is required for Akt-mediated activation of mTORC1, because mutation of serine 2448 to alanine, which prevents its phosphorylation, does not affect downstream signaling to two substrates of mTORC1, S6K1 (p70 ribosomal protein S6 kinase) and 4E-BP1 (eIF4E binding protein 1) [86].
Akt-independent mTOR regulation
There are five pathways for this mechanism of mTOR regulation. mTOR is regulated by mitogenic signals through activation of the Ras/MEK/ERK (guanine triphosphate bindig protein/mitogen-activated protein kinase kinase/extracellular regulated kinase) pathway (Figure 2) changes in the energy status of the cell, in response to hypoxia, by availability of nutrients, such as amino acids and by phospholipase D/phosphatidic acid lipid-signaling cascade [87].
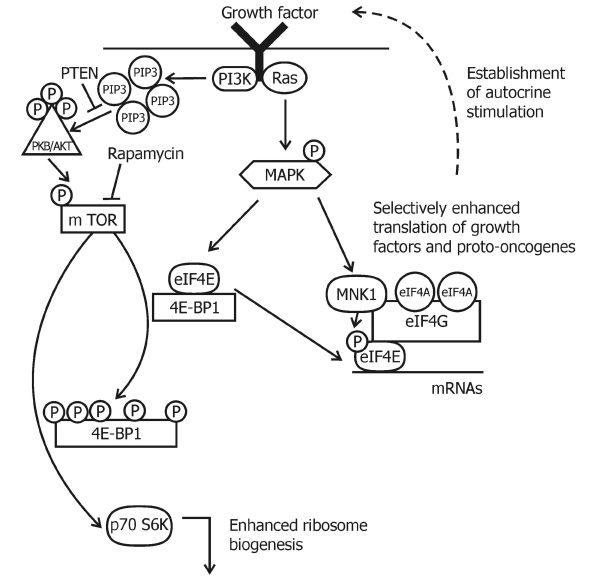
Figure 2: Pharmacological possibilities to inhibit the mTOR signaling pathway via AMPK. AMPK inhibits mTORC1 by direct and indirect mechanisms. Drugs that activate AMPK could be effective in the treatment of cancer. AMPK activators (shown in bold) are metformin, AICAR, 2-DG, PIAs, and A-769662. Metformin is a biguanide widely prescribed for the treatment of the type II diabetes. AICAR is an AMP mimetic, and 2-DG is a glucose analogue. These three drugs activate AMPK by mechanisms that are dependent upon the upstream kinase andtum or suppressor, LKB1. Conversely, the lipid-based Akt inhibitors PIAs, activate AMPK independently of LKB1 (or Akt, not shown). However, cellular activation of AMPK by PIAs is dependent upon another upstream kinase CaMKKβ. The thienopyridone A-769662 activates purified AMPK in vitro, and might also activate AMPK directly in cells.
Inhibition of mTOR in response to low intracellular energy levels is mediated by AMP-activated kinase (AMPK) and its activator (Figure 3), the protein kinase LKB1 [88]. AMPK is a serine-threonine kinase, a heterotrimeric complex of catalytic alpha-subunits and regulatory beta- and gamma-subunits. When intracellular ATP is depleted and the AMP level is increased, AMP binds to AMPK and allows LKB1 to phosphorylate T172 on the catalytic α subunit to activate AMPK [89]. AMPK then phosphorylates TSC2 on S1345, which primes TSC2 for subsequent phosphorylation of S1341 and S1337 by glycogen synthase kinase 3 [90]. GTPase activating protein activity of TSC2 is increased by these phosphorylations and inactivates Rheb, and turns off mTORC1 signaling.
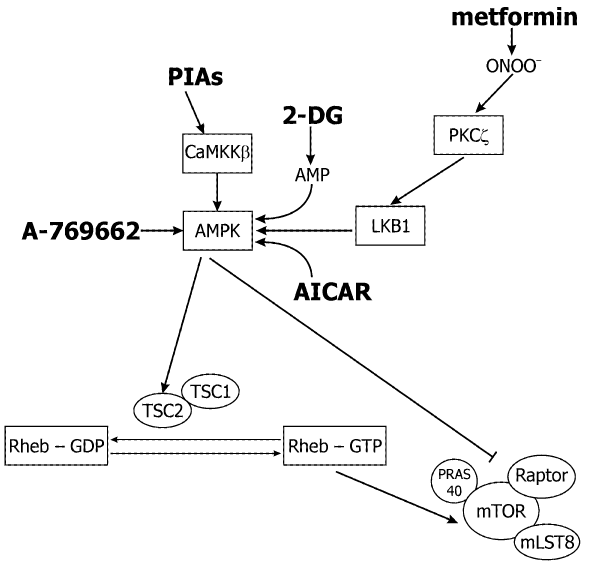
Figure 3: A schematic depiction of mTOR and MAPK signaling in the regulation of translation. mTOR is regulated by multiple inputs, such as for example growth factors, to control translation. Arrows indicate activation and bars indicate inhibition. Activation of PI3K and Ras leads to a cascade of phosphorylation events that results in phosphorylation of the 4E-BP1, its dissociation from eIF4E and an increase in translation. The translational activity of eIF4E can be also modulated by phosphorylation via Mnk1 (MAP kinase-interacting protein kinase-1) which is activated through the Ras-regulated MAPK cascade.
4E-BP family of repressors of mRNA translation
A trimeric complex of eukaryotic initiation factors (eIFs), eIF4F is formed during the initiation phase of mRNA translation. Eukaryotic translation initiation factor 4E (eIF4E) binds to the mRNA cap structure and interacts with the RNA helicase eIF4A and a large scaffold protein eIF4G to create the eIF4F complex. Binding of eIF4E to eIF4G can be blocked by eIF4E-binding proteins (4E-BP1, 4E-BP2 and 4E-BP3) acting as competitive inhibitors of eIF4E-eIF4G interaction [91]. Scaffold protein eIF4G brings the mRNA to the 40 S small ribosomal subunit in a complex with eIF2, GTP and the initiator methionine-transfer RNA by its interaction with eIF3 and forms the 48 S preinitiation complex on the mRNA. Scanning of the mRNA with the aid of eIF4A and recognition of initiation AUG start codon in an optimal context is required and other factors as well as the 60S ribosomal subunit are then recruited and polypeptide chain elongation begins.
The eIF4E activity is controlled by mammalian target of rapamycin (mTOR) through phosphorylation of the eIF4E-binding proteins (4E-BP) [92,93]. mTOR-mediated phosphorylation of 4E-BP liberates eIF-4E enabling assembly of the eIF4F complex. Enhanced eIF4F complex formation increases the translation of all cap-dependent mRNAs and increases protein synthesis rate. eIF4E preferentially enhances translation of a subset of mRNAs, which have lengthy, G and C rich and complex, highly structured 5´ UTRs. These mRNAs are products of expression of transformation-related and survival genes (e.g., c-myc, platelet derived growth factor /PDGF/, vascular endothelial growth factor /VEGF/, insulin-like growth factor 2 /IGF-2/, fibroblast growth factor-2 /FGF-2/, cyclin D1, ornithine decarboxylase /ODC/, survivin and other genes) and their translation is highly sensitive to eIF4E availability [90, 91]. They are poorly translated under normal conditions when eIF4E level is limiting for the eIF4F complex formation. However, most cellular mRNAs have short and unstructured 5´UTR (e.g., housekeeping genes products GAPDH /glyceraldehyde-3-phosphate dehydrogenase/, β-actin and others mRNA) that enable efficient scanning of the 40S ribosomal subunit, initiation codon recognition, second ribosomal subunit loading and translation even when eIF4F complex is limiting.
Ribosomal protein S6 kinase
Mammalian cells contain two similar S6 kinase proteins (S6K1 and S6K2) [94]. S6K1 controls cell growth via increased mRNA translation [94-96]. S6K1 phosphorylates ribosomal protein S6 and enhances the translation of mRNAs possessing a 5´-terminal oligopyrimidine tract (TOP). However, other scientists showed that translation of TOP mRNAs is primarily regulated by growth and mitogenic cues through the PI3-kinase pathway, with a minor role, if any, for the mTOR pathway and ribosomal protein S6 phosphorylation by its kinase, S6K1 [97].
S6K1 also phosphorylates other important targets, including insulin receptor substrate 1 (IRS-1), eukaryotic initiation factor 4B, programmed cell death 4, eukaryotic elongation factor-2 kinase, mTOR, glycogen synthase kinase 3, and other targets [98-100].
Both eIF4E and S6K1 are implicated in cellular transformation, and their overexpression has been linked to poor cancer prognosis [101-106].
Recent evidence suggests that targeting the mTOR pathway may be of clinical/translational value for the treatment of AML, as a majority of patient´s blasts have constitutive activation of Akt with subsequent phosphorylation and activation of down-stream mTOR pathway, including 4E-BP1 and S6K1 [11,14,15].
Table 1 shows clinical trials with rapamycin and related rapamycin analogs (rapalogs) as single agent or in combination with further chemotherapy in AML [107-114]. Rapamycin (Sirolimus) is an immunosuppressive agent, originally developed as an antifungal medicament [115]. Sirolimus is the USAN-assigned generic name for the natural product rapamycin. Sirolimus is produced by a strain of Streptomyces hygroscopicus, isolated from a soil sample collected from Rapa Nui commonly known as Easter Island. Although sirolimus was isolated as an antifungal agent with potent anticandida activity, subsequent studies revealed impressive antitumor and immunosuppressive activities. When tested as a monotherapy in nine patients with reffactory/relapsed AML, four demonstrated a partial response that consisted of reduction in AML blasts by at least 50%. The responses in these four patients were also accompanied by recovering neutrophil counts, and in general the rapamycin was well-tolerated [107]. The rapalogs have better solubility and stability in comparison with sirolimus. They have demonstrated only modest anti-leukemic activity when tested as monotherapies. Deforolimus, a non pro-drug and derivative of rapamycin, was also tested as a monotherapy in AML patients [116]. When deforolimus was given alone to 23 pre-treated AML patients, four displayed stable disease or hematological response, however none of the patients had a complete or even partial response [110].
Table 1: Published clinical trials of rapamycin and its analogs (rapalogs) in AML patients
Clinical trial |
Drug |
Reference |
Rapamycin in 9 refractory, relapsed, or poor-risk AML patients |
Sirolimus |
107 |
Rapamycin in 5 AML patients with no remission after one or multiple chemotherapy cycles |
Sirolimus |
108 |
A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for 28 AML patients |
Everolimus, 3+7 Daunorubicin + Cytarabine |
109 |
A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mTOR inhibitor, in 55 patients with relapsed, refractory hematologic malignancies (23 with AML) |
Deforolimus |
110 |
A phase I study of mTOR inhibitor sirolimus and MEC chemotherapyin relapsed and refractory , or untreated secondary 29 AML patients |
Sirolimus, Mitoxantrone, Etoposide, and Cytarabine (MEC) |
111 |
Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients (60+ y/o) with refractory AML-a phase II GIMEMA study (AML1107) |
Temsirolimus plus Clofarabine (CLO-TOR) |
112 |
Phase I/II study of mTOR inhibitor everolimus in relapsed, refractory, or advanced 27 patients with hematologic malignancies (9 AML patients) |
Everolimus |
113 |
Phase Ib open label dose escalation study in 24 untreated, elderly AML study with everolimus in combination with low dose cytarabine (LDAC) |
Everolimus in combination with low dose Cytarabine (LDAC) |
114 |
Adapted according Calimeri and Ferreri [34] and Herschbein and Liesveld [35]
mTORC1 inhibitors in combination with more traditional forms of chemotherapy were also examined. Everolimus, when combined with daunorubicin and cytarabine in patients experiencing a first relapse had a median overall survival of 19 months and a 68% complete response rate [109]. In patients with relapsed/refractory AML, treatment with sirolimus in combination with mitoxantrone, etoposide, and cytarabine (MEC) had a 22% response, not clearly superior to MEC itself [111]. Treatment with temsirolimus plus clofarabine (CLO-TOR) yielded in 53 elderly AML patients a 21% remission rate with a median overall survival of 4 months overall and 9 months for responders [112].
Phase I/II study of the mTOR inhibitor everolimus (RAD001) was conducted in patients with relapsed or refractory hematologic malignancies (27 patients, 9 AML patients) [113]. Two dose levels (5 and 10 mg orally once daily continuously) were used. None of the nine patients with AML treated with everolimus had an objective response, in contrast to an overall response rate of 44% (44% partial response) reported with sirolimus in patients with AML [107]. It is unclear whether this is due to differences in patient characteristics, duration of therapy (only 28 days with sirolimus), or significant differences in the agents [42]. We observed no responses in 10 patients with AML treated with temsirolimus [117]. On interim analysis, we observed that only 2 of 26 (8%) of patients with AML or MDS treated with AP23573 had a hematologic improvement [118]. In an unfit elderly AML population (24 untreated. elderly AML patients), everolimus in combination with low dose cytarabine (LDAC) followed by everolimus maintenance therapy was tolerable and active, with extended survival observed in those achieving hematologic response [114].
Xu et al. [119] demonstrated that sirolimus enhanced the sensitivity of AML blasts to etoposide in vitro. mTOR inhibition increases sensitivity of AML cells to histone deacetylases inhibitors and to inhibitors of glycolysis [120,121]. Targeting PI3K with LY294002, Akt with perifosine, and both PI3K and mTOR with the dual inhibitor PI-103 have shown promise in preclinical studies [11,122,123]. The dual PI3K/mTOR inhibitor PI-103 prevents p53 induction by mdm2 (murine double minute oncogene and also E3 ubiquitin ligase) inhibition but enhances p53-mediated mitochondrial apoptosis in p53-wild type AML [124]. Rapamycin derivatives reduce not only mTORC1 containing mTOR and raptor with decreased S6K1 and 4E-BP1 phosphorylation, but also blocked Akt activation via inhibition of mTORC2 formation and signaling in AML [70].
VEGF (vascular endothelial growth factor) is a well known regulator of leukemia-associated angiogenesis [125-127] and is also an important paracrine and autocrine growth regulator in AML [125,128-130]. Targeting of mTOR in AML cells by rapamycin and rapamycin derivatives (RAD001 and CCI-779) is associated with decreased growth, decreased survival and downregulation of VEGF [131].
Weisberg et al. [132] investigated the ability of BAG956, a dual PI3K/PDK-1 inhibitor synthesized by Novartis Pharma AG, Basel, Switzerland, to be used in combination with the mTOR inhibitors, rapamycin and its derivative RAD001 (everolimus). They shown that BAG956 and its combination with rapamycin or with RAD001 effective inhibit growth of mutant FLT3-expressing cell lines and AML patient bone marrow cells. Enhancement of the inhibitory effects of tyrosine kinase inhibitor PKC412, by BAG956 and its combination with rapamycin/RAD001 was also shown. The chemical structure of BAG956 is shown in scheme 1 as compound 6 in the article [133]. Further ongoing clinical trials with rapalogs, catalytic inhibitors, and dual inhibitors of PI3K/Akt/mTOR pathway in AML patients were described [27,31-35,38]. Rapalogs are allosteric mTOR inhibitors that bind FKBP12 and mTOR and predominantly inhibit mTORC1. Mechanisms of acquired resistance to rapalogs are unknown. However, an alternate approach is to target mTOR kinase by catalytic inhibitors that potently inhibit mTORC1 as well as mTORC2, thus inhibiting Akt S473 phosphorylation, and thereby preventing or attenuating the feedback loop activation of Akt (Figure 4).
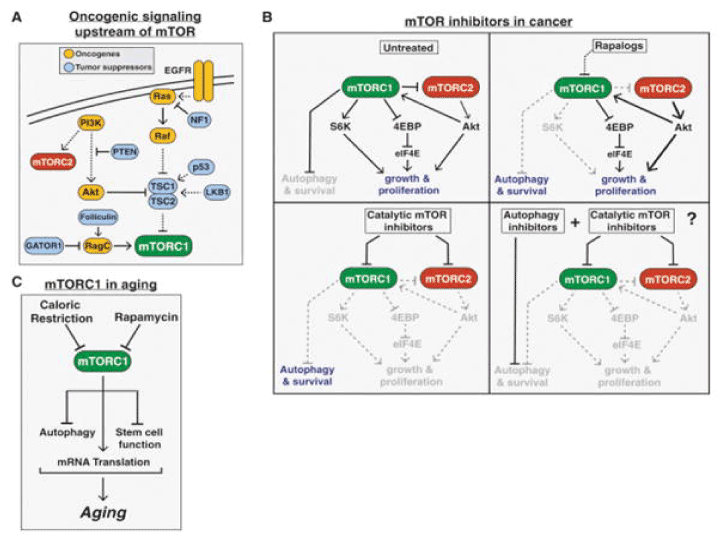
Figure 4: mTOR in cancer and aging. (A) The common tumor suppressors and oncogenes upstream of mTORC1 leading to increased mTORC1 signaling in a wide variety of cancers. (B) The varying effects of rapalogs, catalytic mTOR inhibitors, a combination of an mTOR inhibitor and autophagy inhibitor on cancer proliferation and survival. (C) The role of mTORC1 signaling in aging.
Dual inhibitors of PI3K and mTOR overcome some of the intrinsic disadvantages of rapamycin and its derivatives. Dactolisib (codenamed NVP-BEZ235 and BEZ-235) is an imidazoquinoline derivative acting as a PI3K inhibitor. It also inhibits mTOR. Deng et al. [134] evaluated the effects of BEZ235, a PI3K/mTOR dual inhibitor, on the multidrug-resistant AML cell lines HL-60/VCR (vincristine) and K562/ADR (adriamycin) in vitro. BEZ235 dose-dependently inhibited the viability of HL-60/VCR and K562/ADR cells with the IC50 values of 66.69 and 71.44 nmol/L, respectively. BEZ235 (25–100 nmol/L) dose-dependently inhibited the migration of the two AML cell lines, and it also significantly sensitized the two AML cell lines to VCR and ADR. After treatment with BEZ235, the miR-1-3p levels were markedly increased in HL-60/VCR cells. Using TargetScan analysis and luciferase assays, we showed that miR-1-3p targeted BAG4, EDN1 and ABCB1, the key regulators of cell apoptosis, migration and multidrug resistance, and significantly decreased their levels in the two AML cell lines. Transfection of HL-60/VCR and K562/ADR cells with miR-1-3p-AMO to inhibit miR-1-3p could reverse the anti-proliferation effects of BEZ235. The PI3K/mTOR dual inhibitor BEZ235 effectively chemosensitizes AML cells via increasing miR-1-3p and subsequently down-regulating BAG4, EDN1 and ABCB1. The NVP-BEZ235 compound was found to inhibit PI3K and mTORC1 signaling and also mTORC2 activity [23]. Furthermore, NVP-BEZ235 fully inhibits the rapamycin-resistant phosphorylation of 4E-BP1, resulting in a marked inhibition of protein translation in AML cells. Therefore, NVP-BEZ235 reduces the proliferation rate and induces an important apoptotic response in AML cells without affecting normal CD34(+) survival.
NVP-BEZ235 was substantially more effective in inhibiting PI3K and Akt activity, when compared to rapamycin. This was confirmed in AML cell lines MV4-11, MOLM-14, and OCI-AML3 and in bone marrow samples taken from patients recently diagnosed with AML [23]. Complete dephosphorylation of 4E-BP1 resulted in the reduction of cap-dependent protein translation, specifically of oncogenic proteins c-Myc, cyclin D1, and Bcl-xL [135]. NVP-BEZ235 inhibited also cell cycling. Exposed AML cell lines had an increase in the proportion of cells in the G0/G1 phase and a decrease of cells in the S and G/M phases of the cell cyle.
NVPBGT226 is a dual PI3K/mTOR inhibitor has very similar action to NVP-BEZ235. AML with mutated FLT3 and KIT were treated with NVPBGT226. This inhibitor was able to suppress PI3K, Akt, and mTORC1/2 activity. NVPBGT226 caused potent induction of apoptosis, as opposed to the G0/G1 arrest seen with NVP-BEZ235 [136].
Gedatolisib (PKI-587), apitolisib (GDC-0980), BGT226, and dactolisib showed similar responses in 25 AML cell lines, where part of these cell lines was sensitive and part unsensitive with substantial resistance to these dual PI3K/mTOR inhibitors [137]. Using RNA sequencing data, it was found that expression of a G protein-coupled receptor for uridine diphosphate glucose, P2RY14 (also known as GPR105), was upregulated nine-fold in cells showing resistance to the PI3K/mTOR inhibitor. P2RY14 has not been much studied in hematologic malignancies. However, this receptor seems to have a role in the localization of hematopoietic stem cells (HSCs) and in promoting regenerative capabilities following injury. GPR105 identifies a quiescent, primitive population of hematopoietic cells restricted to bone marrow. It mediates primitive cell responses to specific hematopoietic microenvironments and extends the known immune system functions of P2Y receptors to the stem cell level [138]. GPR105 provides also a novel link between innate immunity and metabolism [139].
The tricyclic purine nucleoside analog triciribine mono-phoshate monohydrate (TCN-PM) inhibits Akt phosphorylation by interfering with Akt´s PH domain and preventing its membrane localization [140]. Triciribine was evaluated in 41 patients with advanced hematologic malignancies, including 36 AML patients. The toxicity of the treatment was acceptable and a significant reduction in the levels of phosphorylated Akt (S473) and BAD (S112) was found. TCN-PM therapy was well tolerated in patients with advanced hematological malignancies, and reduced levels of phosphorylation of Akt and its substrate Bad were consistent with inhibition of this PI3K/Akt/mTOR survival pathway and induction of cell death.
MK-2206 is an oral non-ATP competitive allosteric inhibitor of Akt and induces apoptosis and cell cycle arrest of AML cell lines. However, it demonstrated limited efficacy during a phase I study with only one response among 18 patients (complete remission with incomplete platelet count recovery), leading to early termination of the trial [141]. The most common grade 3/4 drug-related toxicity was a pruritic rash in 6 of 18 patients. Nevertheless, despite the use of MK-2206 at maximum tolerated doses, reverse phase protein arrays (RPPA) analyses indicated only modest decreases in Ser473 AKT (median 28%; range, 12%-45%) and limited inhibition of downstream targets.
UCN-01 and perifosine inhibit Akt activation. UCN-01 is a synthetic derivative of staurosporine (7-hydroxystaurosporine) with antiproliferative activity against several in vitro and in vivo cancer models. The primary objective of the phase I study was to determine the maximum tolerated dose (MTD) of UCN-01 given in combination with perifosine in patients with advanced acute leukemias and myelodysplastic syndrome [142]. Secondary objectives included safety, pharmacokinetics, pharmacodynamics, and efficacy. Perifosine 150 mg every 6 h was given orally on day 1 followed by 100 mg once a day continuously in 28-day cycles. UCN-01 was given intravenously over 3 h on day 4 at three dose levels (40 mg/m2, 65 mg/m2, 90 mg/m2). UCN-01 and perifosine can be safely administered, but this regimen lacked clinical efficacy. This approach may have failed because of insufficient Akt inhibition in vivo.
PIM kinases (PIM1, 2 and 3) are involved in cell proliferation and survival signalling and are emerging targets for the therapy of various malignancies. Meja et al. [143] found that a significant proportion of primary acute myeloid leukaemia (AML) samples showed PIM1 and PIM2 expression by quantitative reverse transcription polymerase chain reaction. Therefore, we investigated the effects of a novel ATP-competitive pan-PIM inhibitor, AZD1897, on AML cell growth and survival. PIM inhibition showed limited single agent activity in AML cell lines and primary AML cells, including those with or without FLT3-internal tandem duplication (ITD) mutation. However, significant synergy was seen when AZD1897 was combined with the Akt inhibitor AZD5363, a compound that is in early-phase clinical trials. AML cells from putative leukaemia stem cell subsets, including CD34+38- and CD34+38+ fractions, were equivalently affected by dual PIM/Akt inhibition when compared with bulk tumour cells. Analysis of downstream signalling pathways showed that combined PIM/Akt inhibition downregulated mTOR outputs (phosphorylation of 4EBP1 and S6) and markedly reduced levels of the anti-apoptotic protein MCL1. The combination of PIM and Akt inhibition holds promise for the treatment of AML.
Inhibitors of phosphoinositide 3-kinase delta
Duvelisib (Copiktra®) is a dual inhibitor of phosphoinositide 3-kinases (PI3Kδ and PI3Kγ). In 2018, duvelisib was first approved by the FDA for the treatment of adult patients with relapsed or refractory chronic lymphocytic leukaemia (CLL)/ small lymphocytic lymphoma (SLL) after at least two prior therapies [144]. Duvelisib has also been approved under accelerated track for relapsed or refractory follicular lymphoma (FL) after at least two prior systemic therapies.
The PI3Kδ isoform is always expressed in AML cells, whereas the frequency of PI3Kγ expression is highly variable. The functions of these individual catalytic enzymes have not been fully resolved in AML, therefore using the PI3K p110δ and p110γ-targeted inhibitor IPI-145 (duvelisib) and specific p110δ and p110γ shRNA, Pillinger et al. [145] analysed the role of these two p110 subunits in human AML blast survival. The results showed that PI3Kδ and PI3Kγ inhibition with IPI-145 had anti-proliferative activity in primary AML cells by inhibiting the activity of AKT and mitogen-activated protein kinase (MAPK). Pre-treatment of AML cells with IPI-145 inhibited both adhesion and migration of AML blasts to bone marrow stromal cells (BMSCs). Using shRNA targeted to the individual isoforms they demonstrated that p110δ-knockdown had a more significant anti-proliferative effect on AML cells, whereas targeting p110γ-knockdown significantly inhibited AML migration. The results demonstrated that targeting both PI3Kδ and PI3Kγ to inhibit AML-BMSC interactions provides a biologic rationale for the pre-clinical evaluation of IPI-145 in AML.
A medicinal chemistry effort focused on identifying a structurally diverse candidate for PI3Kd led to the discovery of clinical candidate INCB050465 (20, parsaclisib). The unique structure of 20 contains a pyrazolopyrimidine hinge-binder in place of a purine motif that is present in other PI3Kd inhibitors, such as idelalisib (1), duvelisib (2), and INCB040093 (dezapelisib) (3). Parsaclisib (20) is a potent and highly selective inhibitor of PI3Kd (Figure 5) with drug-like ADME (absorption, distribution, metabolism, and excretion) properties that exhibited an excellent in vivo profile as demonstrated through pharmacokinetic studies in rats, dogs, and monkeys and through pharmacodynamic and efficacy studies in a mouse Pfeiffer EZH2 A682G mutant xenograft model [146].
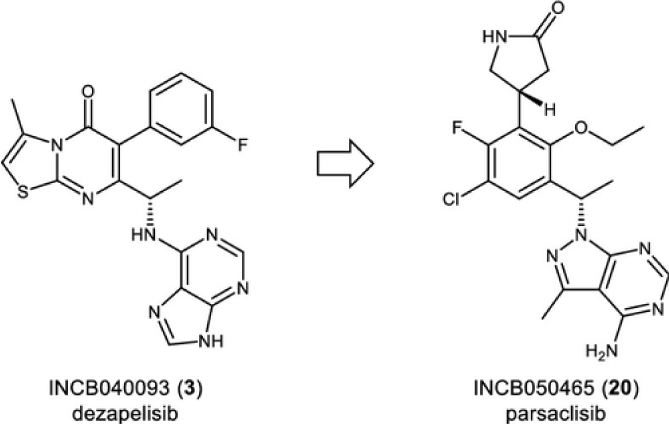
Figure 5: Chemical structure of phosphoinositide 3-kinase delta (PI3Kδ) inhibitors dezapelisib (INCB040093) (3) and parsaclisib (INCB050465) [20].
PI3Kd was detected in all 14 studied cases of AML whereas the expression levels of the other class I PI3Ks varied more widely, and were often undetectable [147]. The p110δ-selective compound IC87114 inhibited constitutive phosphorylation of the PI3K target Akt/PKB and reduced cell number to a mean of 66% (range 14 - 88%). In eight cases, the combination of IC87114 and VP16 (a topoisomerase II inhibitor) was synergistic in reducing viable cell number, and was associated with a reduction in constitutive NF-κB activity. IC87114 did not have direct adverse effects or enhance the activity of VP16 on the proliferation and survival of normal haemopoietic progenitors [147]. Overall, these results identified the p110δ isoform as a potential therapeutic target in AML and supported a clinical approach to use isoform-selective over broad-spectrum PI3K inhibitors.
Several research groups have shown that a high level mTOR expression is able to control apoptosis by modulating several molecules, including Bcl-2 family members, and thus promoting tumor cell survival [148].
Inhibitors targeting BCL-2 apoptotic proteins have significant potential for the treatment of AML. However, complete responses are observed in only 20% of patients, suggesting that targeting BCL-2 alone is insufficient to yield durable responses. Rahmani et al. [149] assessed the efficacy of coadministration of the PI3K/mTOR inhibitor GDC-0980 or the p110β-sparing PI3K inhibitor taselisib with the selective BCL-2 antagonist venetoclax in AML cells. Tetracycline-inducible downregulation of BCL-2 significantly sensitized MV4-11 and MOLM-13 AML cells to PI3K inhibition. Venetoclax/GDC-0980 coadministration induced rapid and pronounced BAX mitochondrial translocation, cytochrome c release, and apoptosis in various AML cell lines in association with AKT/mTOR inactivation, and MCL-1 downregulation. Ectopic expression of MCL-1 significantly protected cells from this regimen. Combined treatment was also effective against primary AML blasts from 17 patients, including those bearing various genetic abnormalities. Venetoclax/GDC-0980 markedly induced apoptosis in primitive CD34+/38−/123+ AML cell populations but not in normal hematopoietic progenitor CD34+ cells. The regimen was also active against AML cells displaying intrinsic or acquired venetoclax resistance or tumor microenvironment–associated resistance. Either combinatorial treatment markedly reduced AML growth and prolonged survival in a systemic AML xenograft mouse model and diminished AML growth in two patient-derived xenograft models. Venetoclax/GDC-0980 activity was partially diminished in BAK−/− cells and failed to induce apoptosis in BAX−/− and BAX−/−BAK−/− cells, whereas BIM−/− cells were fully sensitive. Similar results were observed with venetoclax alone in in vitro and in vivo systemic xenograft models. Collectively, these studies demonstrated that venetoclax/GDC-0980 exhibited potent anti-AML activity primarily through BAX and, to a lesser extent, BAK [149]. These findings argued that dual BCL-2 and PI3K inhibition warrants further evaluation in AML.
Effects of concurrent inhibition of mTORC1/2 and Bcl-2/Bcl-xL in human acute myeloid leukemia cells were examined [150]. Tetracycline-inducible Bcl-2/Bcl-xL dual knockdown markedly sensitized acute myeloid leukemia cells to the dual TORC1/2 inhibitor INK128 in vitro as well as in vivo. Moreover, INK128 co-administered with the Bcl-2/xL antagonist ABT-737 sharply induced cell death in multiple acute myeloid leukemia cell lines, including TKI-resistant FLT3-ITD mutants and primary acute myeloid leukemia blasts carrying various genetic aberrations, e.g., FLT3, IDH2, NPM1, and Kras, while exerting minimal toxicity toward normal hematopoietic CD34+ cells. Combined treatment was particularly active against CD34+/CD38−/CD123+ primitive leukemic progenitor cells. The INK128/ABT-737 regimen was also effective in the presence of a protective stromal microenvironment. Notably, INK128 was more potent than the TORC1 inhibitor rapamycin in down-regulating Mcl-1, diminishing AKT and 4EBP1 phosphorylation, and potentiating ABT-737 activity. Mcl-1 ectopic expression dramatically attenuated INK128/ABT-737 lethality, indicating an important functional role for Mcl-1 down-regulation in INK128/ABT-737 actions. Immunoprecipitation analysis revealed that combined treatment markedly diminished Bax, Bak, and Bim binding to all major anti-apoptotic Bcl-2 members (Bcl-2/Bcl-xL/Mcl-1), while Bax/Bak knockdown reduced cell death. Finally, INK128/ABT-737 co-administration sharply attenuated leukemia growth and significantly prolonged survival in a systemic acute myeloid leukemia xenograft model. Analysis of subcutaneous AML-derived tumors revealed significant decrease in 4EBP1 phosphorylation and Mcl-1 protein level, consistent with results obtained in vitro. These findings demonstrate that co-administration of dual mTORC1/mTORC2 inhibitors and BH3-mimetics exhibits potent anti-leukemic activity in vitro and in vivo [150]. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhanced PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism [151].
Synergistic effects of dual PI3K/mTOR inhibitor and MEK inhibition on apoptosis
The MAPK signaling pathways involve a family of protein kinases that play critical roles in regulation of diverse cellular activities, including cell proliferation, survival, differentiation, motility, and angiogenesis. The MAPK pathways transduce signals from various extracellular stimuli (growth factors, hormones, cytokines and environmental stresses), leading to distinct intracellular responses via a series of phosphorylation events and protein-protein interactions [152]. Four distinct MAPK cascades have been identified and named according to their MAPK module. These are extracellular signal-regulated kinase (ERK1/2), c-Jun N-terminal kinase (JNK), p38 and ERK5. Each of these cascades comprised of three sequentially acting kinases, activating one after the other (MAPKKK/MAP3K, MAPKK/MAP2K, and MAPK). These signaling cascades are often dysregulated in human cancer cells [26]. Many small molecule inhibitors targeting various component of these cascades were developed. Mitogen-activated protein kinase or MAP2K or MAPKK are commonly known as MEK proteins.
In MLL-rearranged THP-1cells and patient-derived xenografted (PDX) cells, a combination treatment of the dual PI3K/mTOR inhibitor BEZ235 with an MEK inhibitor AZD-6224 showed highly synergistic effects on apoptosis [28]. When THP-1 cells were treated with BEZ235 for 24 h, an increase of ERK phosphorylation was found. Treatment of THP-1 cells with the MEK inhibitor AZD-6224 showed increased Akt phosphorylation. This was further confirmed in PDX cells, and in MOLM-13 and HL60 cells and was not specific for MLL-rearranged or RAS-mutated cells.
Toxicities observed with mTOR inhibitors consisted of hyperglycemia, hyperlipidemia, anorexia, elevated liver enzymes, oral aphthous ulcers, diarrhea, hypophosphatemia, fatigue, and hypomagnesemia. Inhibitors of mTOR are safe and well-tolerated. PI3K and its downstream effector Akt which contributes to mTOR phosphorylation and activation are abberantly activated in many hematological malignancies. Therefore, PI3k/Akt/mTOR signaling pathway is promising target in the treatment of hematological malignancies, including AML, especially by using of combinations of mTOR inhibitors with conventional cytotoxic agents.
Even through constitutive activation of PI3K/Akt/mTOR signaling in the enriched leukemic cells is seen for most AML patients, resistance to the antiproliferative effect of pathway inhibitors is relatively common.Leukemia cells from resistant patient differ with regard to their metabolomic profile, including the metabolites involved in amino acid and arachidonic acid metabolism. Arachidonic acid metabolism is important for survival and proliferation of hematopoietic cells. Several arachidonic acid metabolites (prostaglandins and eicosatetraenoic acid derivatives) can influence activation and signaling through the PI3K/Akt/mTOR pathway [38]. Insulin is a regulator of cellular proliferation, survival, and metabolism. The PI3K/Akt/mTOR pathway is one of several pathways that are involved in regulation of these cellular functions and which are regulated by insulin and insulin receptors [40,75,78,82-85], The heterogeneity of AML patients is seen not only in their responsiveness to the PI3K/Akt/mTOR pathway inhibitors but also to insulin. Insulin is present in the bone marrow microenvironment. AML patients can be subclassified based on the effect of insulin on the phosphorylation of the PI3K/Akt/mTOR pathway mediators, and these subsets differ in their global geneexpression as well as their proteomic and phosphoproteomic profiles. The subset with the highest PI3K/Akt/mTOR pathway activation in response to insulin also showed weakened antileukemic effects of metabolic inhibitors. Variations between primary AML cells their communication with leukemia-supporting neighbouring cells in the bone marrow microenvironment will also differ. Direct effects of the PI3K/Akt/mTOR pathway inhibition on the AML cells communication with leukemia-supporting neighbouring cells in the bone marrow microenvironment will thereby be difficult to predict and may even increase the patient heterogeneity to the final effect of the treatment [31].
It is rather unlikely that inhibition of the PI3K/Akt/mTOR pathway as a single approach will be effective due to feedback mechanisms and development of resistance. Numerous other mechanisms are often concurrently active to support proliferation and survival advantage to AML progenitors and stem cells. These include epigenetic modulation, cell cycle activation, signal transduction pathway activation, anti-apoptotic mediator expression, and abnormalities of oxidative phosphorylation, autophagy, and proteasomal/neddylation pathways, amongst others. Therefore, it is likely that success of the PI3K/Akt/mTOR pathway inhibition in AML will require combination with inhibitors of other pathways or with standard cytotoxic chemotherapy procedures [28,35,149-151,153].
This work was supported by the project for conceptual development of research organization No 00023736 (Institute of Hematology and Blood Transfusion) from the Ministry of Health of the Czech Republic.
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, et al. (1994) A mammalian protein targeted by G1 –arresting rapamycin – receptor complex. Nature 369: 756-758. [Crossref]
- Chiu MI, Katz H, Berlin V (1994) RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci U S A 91: 12574-12578. [Crossref]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH (1994) RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin- dependent fashion and is homologous to yeast TORs. Cell 78: 35-43. [Crossref]
- LoPiccolo J1, Blumenthal GM, Bernstein WB, Dennis PA (2008) Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat 11: 32-50. [Crossref]
- Memmott RM, Dennis PA (2009) Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal 21: 656-664. [Crossref]
- Baldo P, Cecco S, Giacomin E, Lazzarini R, Ros B, et al. (2008) mTOR pathway and mTOR inhibitors as agents for cancer therapy. Curr Cancer Drug Targets 8: 647-665. [Crossref]
- Hudes GR (2009) Targeting mTOR in renal cell carcinoma. Cancer 115: 2313-2320. [Crossref]
- Malizia LJ, Hsu A (2008) Temsirolimus, an mTOR inhibitor for treatment of patients with advanced renal carcinoma. Clin J Oncol Nurs 12: 639-646. [Crossref]
- Le Tourneau C, Faivre S, Serova M, Raymond E (2008) mTORC1 inhibitors: is temsirolimus in renal cancer telling us how they really work. Br J Cancer 99: 1197-1203. [Crossref]
- Dasanu CA, Clark BA 3rd, Alexandrescu DT (2009) mTOR-blocking agents in advanced renal cancer: an emerging therapeutic option. Expert Opin Investig Drugs 18: 175-187. [Crossref]
- Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M (2003) Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 102: 972-980. [Crossref]
- Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, et al. (2003) Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia 17: 993-997. [Crossref]
- Zhao S, Konopleva M, Cabreira-Hansen M, Xie Z, Hu W, et al. (2004) Inhibition of phosphatidylinositol 3-kinase dephosphorylates BAD and promotes apoptosis in myeloid leukemias. Leukemia 18: 267-275. [Crossref]
- Récher C, Dos Santos C, Demur C, Payrastre B (2005) mTOR, a new therapeutic target in acute myeloid leukemia. Cell Cycle 4: 1540-1549. [Crossref]
- Récher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, et al. (2005) Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 105: 2527-2534. [Crossref]
- Grandage VL, Gale RE, Linch DC, Khwaja A (2005) PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, MAP kinase and p53 pathways. Leukemia 19: 586-594. [Crossref]
- Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, et al. (2005) Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 106: 1063-1066. [Crossref]
- Bardet V, Tamburini J, Ifrah N, Dreyfus F, Mayeux P, et al. (2006) Single cell analysis of phosphoinositide 3-kinase/Akt and ERK activation in acute myeloid leukemia by flow cytometry. Haematologica 91: 757-764. [Crossref]
- Kornblau SM1, Womble M, Qiu YH, Jackson CE, Chen W, et al. (2006) Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 108: 2358-2365. [Crossref]
- Tamburini J, Elie C, Bardet V, Chapuis N, Park S, et al. (2007) Constitutive phosphoinositide 3-kinase / Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood 110: 1025-1028. [Crossref]
- Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Manzoli L, et al. (2009) Targeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemia. Expert Opin Investig Drugs 18: 1333-1349. [Crossref]
- Martelli AM, Evangelisti C, Chiarini F, McCubrey JA (2010) The phosphatidylinositol 3-kinase/ Akt/ mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 1: 89-103. [Crossref]
- Chapuis N1, Tamburini J, Green AS, Vignon C, Bardet V, et al. (2010) Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res 16: 5424-5435. [Crossref]
- Mise J1, Dembitz V, Banfic H, Visnjic D (2011) Combined inhibition of PI3K and mTOR exerts synergistic antiproliferative effect, but diminishes differentiative properties of rapamycin in acute myeloid leukemia cells. Pathol Oncol Res 17: 645-656. [Crossref]
- Evangelisti C, Evangelisti C, Bressanin D, Buontempo F, Chiarini F, et al. (2013) Targeting phosphatidylinositol 3-kinase signaling in acute myelogenous leukemia. Expert Opin Ther Targets 17: 921-936. [Crossref]
- Prijić S, Ugrina I, Labar B, Nemet D, Batinić J, et al. (2015) Prognostic significance of constutitive phosphatidylinositol 3-kinase/ Akt/ and mitogen-activated protein kinase phosphorylation in acute myeloid leukemia. Leuk Lymphoma 56: 2281-2288. [Crossref]
- Carneiro BA, Kaplan JB, Altman JK, Giles FJ, Platanias LC (2015) Targeting mTOR signaling pathways and related negative feedback loops for the treatment of acute myeloid leukemia. Cancer Biol Ther 16: 648-656. [Crossref]
- Sandhöfer N, Metzeler KH, Rothenberg M, Herold T, Tiedt S, et al. (2015) Dual PI3K/ mTOR inhibition shows antileukemic activity in MLL-rearranged acute myeloid leukemia. Leukemia 29: 828-838. [Crossref]
- Hauge M, Bruserud Ø, Hatfield KJ (2015) Targeting of cell metabolism in human acute myeloid leukemia-more than targeting of isocitrate dehydrogenase mutations and PI3K/ AKT/ mTOR signaling? Eur J Haematol 96: 211-221. [Crossref]
- Lindblad O, Cordero E, Puissant A, Macaulay L, Ramos A, et al. (2016) Aberrant activation of the PI3K/ mTOR pathway promotes resistance to sorafenib in AML. Oncogene 35: 5119-5131. [Crossref]
- Brenner AK, Andersson Tvedt TH, Bruserud Ø (2016) The complexity of targeting PI3K-Akt-mTOR signalling in human acute myeloid leukaemia: The importance of leukemic cell heterogeneity, neighbouring mesenchymal stem cells and immunocompetent cells. Molecules 21: 1512. [Crossref]
- Tabe Y, Tafuri A, Sekihara K, Yang H, Konopleva M (2017) Inhibition of mTOR kinase as a therapeutic target for acute myeloid leukemia. Expert Opin Ther Targets 21: 705-714. [Crossref]
- Ricciardi MR, Mirabilii S, Licchetta R, Piedimonte M, Tafuri A (2017) Targeting the Akt, GSK-3, Bcl-2 axis in acute myeloid leukemia. Adv Biol Regul 65: 36-58. [Crossref]
- Calimeri T, Ferreri AJM (2017) m-TOR inhibitors and their potential role in haematological malignancies. Brit J Haematol 177: 684-702. [Crossref]
- Herschbein L, Liesveld JL (2018) Dueling for dual inhibition: Means to enhance effectiveness of PI3K/ Akt/ mTOR inhibitors in AML. Blood Rev 32: 235-248. [Crossref]
- Nepstad I, Hatfield KJ, Tvedt THA, Reikvam H, Bruserud Ø (2018) Clonal heterogeneity reflected by PI3K-AKT-mTOR signaling in human acute myeloid leukemia cells and nits association with adverse prognosis. Cancers (Basel) 10: 332.
- Nepstad I, Hatfield KJ, Aasebø E, Hernandez-Valladares M, Brenner AK, et al. (2018) Two acute myeloid leukemia patient subsets are identified based on the constitutive PI3K-Akt-mTOR signaling of their leukemic cells; a functional, proteomic, and transcriptomic comparison. Expert Opin Ther Targets 22: 639-653. [Crossref]
- Nepstad I, Reikvam H, Brenner AK, Bruserud Ø, Hatfield KJ (2018) Resistance to the antiproliferative in vitro effect of PI3K-Akt-mTOR inhibition in primary human acute myeloid leukemia cells is associated with altered cell metabolism. Int J Mol Sci 19: 382. [Crossref]
- Bertacchini J, Frasson C, Chiarini F, D'Avella D, Accordi B, et al. (2018) Dual inhibition of PI3K/mTOR signaling in chemoresistant AML primary cells. Adv Biol Regul 68: 2-9. [Crossref]
- Nepstad I, Hatfield KJ, Grǿnningsaeter IS, Aasebø E, Hernandez-Valladares M, et al. (2019) Effects of insulin and pathway inhibitors oén the PI3K-Akt-mTOR phosphorylation profile in acute myeloid leukemia cells. Signal Transduct Target Ther 4: 20.
- Deb G, Wingelhofer B, Amaral FMR, Maiques-Diaz A, Chadwick JA, et al. (2020) Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. Leukemia 34: 1266-1277. [Crossref]
- Panwalkar A, Verstovsek S, Giles FJ.(2004) Mammalian target of rapamycin inhibition as therapy for hematologic malignancies. Cancer 100: 657-666. [Crossref]
- Altman JK, Platanias LC (2008) Exploiting the mammalian target of rapamycin pathway in hematologic malignancies. Curr Opin Hematol 15: 88-94. [Crossref]
- Teachey DT, Grupp SA, Brown VI (2009) Mammalian target of rapamycin inhibitors and their potentional role in therapy in leukaemia and other hematological malignancies. Br J Haematol 145: 569-580. [Crossref]
- Majchrzak A, Witkowska M, Smolewski P (2014) Inhibition of the PI3K/ Akt/ mTOR signaling pathway in diffuse large B-cell lymphoma: current knowledge and clinical significance. Molecules 19: 14304-14315. [Crossref]
- Yang Q, Modi P, Newcomb T, Quéva C, Gandhi V (2015) Idelalisib, first-in-class PI3Kδ inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res 21: 1537-1542. [Crossref]
- Seiler T, Hutter G, Dreyling M (2016) The emerging role of PI3K inhibitors in the treatment of hematological malignancies: preclinical data and clinical progress to date. Drugs 76: 639-646. [Crossref]
- Lampson BL, Brown JR (2017) PI3Kδ-selective and PI3Kα/δ-combinatorial inhibitors in clinical development for B-cell and non-Hodgkin lymphoma. Expert Opin Investig Drugs 26: 1267-1279. [Crossref]
- Erdmann T, Klener P, Lynch JT, Grau M, Vočková P, et al. (2017) Sensitivity to PI3K and AKT inhibitors is mediated by divergent molecular mechanisms in subtypes of DLBCL. Blood 130: 310-322. [Crossref]
- Dreyling M, Santoro A, Mollica L, Leppä S, Follows GA, et al. (2017) Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed a refractory indolent lymphoma. J Clin Oncol 35: 3898-3905. [Crossref]
- von Keudell G, Moskowitz AJ (2019) The role of PI3K inhibition in lymphoid malignancies. Curr Hematol Malig Rep 14: 405-413. [Crossref]
- Wullenkord R, Friedrichs B, Erdmann T, Lenz G (2019) Therapeutic potential of PI3K signaling in distinct intities of B-cell lymphoma. Expert Rev Hematol 12: 1053-1062. [Crossref]
- Simioni C, Martelli AM, Zauli G, Melloni E, Neri LM (2019) Targeting mTOR in acute lymphoblastic leukemia. Cells 8: 190. [Crossref]
- Kienle DL, Stilgenbauer S (2020) Approved and emerging PI3K inhibitors for the treatment of chronic lymphocytic leukemia and non-Hodgkin lymphoma. Expert Opin Pharmacother 12: 1-13. [Crossref]
- Bosotti R, Isacchi A, Sonnhammer EL (2000) FAT: A novel domain in PIK-related kinases. Trends Biochem Sci 25: 225-227. [Crossref]
- Harding MW (2003) Immunophilins, mTOR, and pharmacodynamic strategies for a targeted cancer therapy. Clin Cancer Res 9: 2882-2886. [Crossref]
- Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18: 1926-1945. [Crossref]
- Gunn RM, Hailes HC (2008) Insights into the PI3-K-PKB-mTOR signalling pathway from small molecules. J Chem Biol 1: 49-62. [Crossref]
- Vogt PK, Gymnopoulos M, Hart JR (2009) PI3-kinase and cancer: changing accents. Curr Opin Genet Dev 19: 12-17. [Crossref]
- Engelman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7: 606-619. [Crossref]
- Anderson KE, Jackson SP (2003) Class I phosphoinositide 3-kinases. Int J Biochem Cell Biol 35: 1028-1033. [Crossref]
- Salmena L, Carracedo A, Pandolfi PP (2008) Tenets of PTEN tumor suppression. Cell 133: 403-414. [Crossref]
- Carracedo A, Pandolfi PP (2008) The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27: 5527-5541. [Crossref]
- Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, et al. (1997) High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol Chem 272: 8474-8481. [Crossref]
- Andjelković M, Alessi DR, Meier R, Fernandez A, Lamb NJ, et al. (1997) Role of translocation in the activation and function of protein kinase B. J Biol Chem 272: 31515-31524. [Crossref]
- Anderson KE, Coadwell J, Stephens LR, Hawkins PT (1998) Translocation of PDK-1 to the plasma membrane is important in allowing PDK-1 to activate protein kinase B. Curr Biol 8: 684-691. [Crossref]
- Lim MA, Kikani CK, Wick MJ, Dong LQ (2003) Nuclear translocation of 3´-phosphoinositide-dependent protein kinase 1 (PDK-1): A potential regulatory mechanism for PDK-1 function. Proc Natl Acad Sci USA 100: 14006-14011.
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098-1101. [Crossref]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, et al. (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22: 159-168. [Crossref]
- Zeng Z, Sarbassov dos D, Samudio IJ, Yee KW, Munsell MF, et al. (2007) Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 109: 3509-3512. [Crossref]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648-657. [Crossref]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10: 151-162. [Crossref]
- Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, et al. (2002) Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA 99: 13571-13576. [Crossref]
- Huang J, Manning BD (2008) The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 412: 179-190. [Crossref]
- Avruch J, Hara K, Lin Y, Liu M, Long X, et al. (2006) Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 25: 6361-6372. [Crossref]
- Wang X, Fonseca BD, Tang H, Liu R, Elia A, et al. (2008) Re-evaluating the roles of proposed modulators of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem 283: 30482-30492. [Crossref]
- Bai X, Ma D, Liu A, Shen X, Wang QJ, et al. (2007) Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 318: 977-980. [Crossref]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, et al. (2003) Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 11: 1457-1466. [Crossref]
- Wang L, Lawrence JC Jr, Sturgill TW, Harris TE (2009) Mammalian target of rapamycin complex 1 (mTORC1) activity is associated with phosphorylation of raptor by mTOR. J Biol Chem 284: 14693-14697. [Crossref]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, et al. (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10: 457–468. [Crossref]
- Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, et al. (2003) GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between Raptor and mTOR. Mol Cell 11: 895–904. [Crossref]
- Rodgers BD1, Levine MA, Bernier M, Montrose-Rafizadeh C (2001) Insulin regulation of a novel WD-40 repeat protein in adipocytes. J Endocrinol 168: 325–332. [Crossref]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, et al. (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25: 903-915. [Crossref]
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9: 316-323. [Crossref]
- Navé BT1, Ouwens M, Withers DJ, Alessi DR, Shepherd PR (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J 344: 427-431. [Crossref]
- Sekulić A, Hudson CC, Homme JL, Yin P, Otterness DM, et al. (2000) A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 60: 3504-3513. [Crossref]
- Memmott RM, Dennis PA (2009) Akt-dependent and –independent mechanisms of mTOR regulation in cancer. Cell Signal 21: 656-664. [Crossref]
- Alessi DR, Sakamoto K, Bayascas JR (2006) LKB1-dependent signaling pathways. Annu Rev Biochem 75: 137-163. [Crossref]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, et al. (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 101: 3329-3335. [Crossref]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, et al. (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126: 955-968. [Crossref]
- Goodfellow IG, Roberts LO (2008) Eukaryotic initiation factor 4E. Int J Biochem Cell Biol 40: 2675-2680. [Crossref]
- Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, et al. (2007) Dissecting eIF4E action in tumorigenesis. Genes Dev 21: 3232-3237. [Crossref]
- Graff JR, Konicek BW, Carter JH, Marcusson EG (2008) Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res 68: 631-634. [Crossref]
- Dufner A, Thomas G (1999) Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 1999, 253, 100-109. [Crossref]
- Reiter AK, Bolster DR, Crozier SJ, Kimball SR, Jefferson LS (2008) AMPK represses TOP mRNA translation but not global protein synthesis in liver. Biochem Biophys Res Commun 374: 345-350. [Crossref]
- Meric-Bernstam F, Gonzalez-Angulo AM (2009) Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 27: 2278-2287. [Crossref]
- Stolovich M1, Tang H, Hornstein E, Levy G, Cohen R, et al. (2002) Transduction of growth or mitogenic signals into translational activation of TOP mRNAs is fully reliant on the phosphatidylinositol 3-kinase-mediated pathway but requires neither S6K1 nor rpS6 phosphorylation. Mol Cell Biol 22: 8101-8113. [Crossref]
- Smyk A, Szuminska M, Uniewicz KA, Graves LM, Kozlowski P (2006) Human enhancer of rudimentary is a molecular partner of PDIP46/SKAR, a protein interacting with DNA polymerase delta and S6K1 and regulating cell growth. FEBS J 273: 4728-4741. [Crossref]
- Jastrzebski K, Hannan KM, Tchoubrieva EB, Hannan RD, Pearson RB (2007) Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 25: 209-226. [Crossref]
- Ma XM, Blenis J (2009) Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10: 307-318. [Crossref]
- De Benedetti A, Harris AL (1999) eIF4E expression in tumors: its possible role in progression of malignancies. Int J Biochem Cell Biol 31: 59-72. [Crossref]
- Sorrells DL, Meschonat C, Black D, Li BD (1999) Pattern of amplification and overexpression of the eukaryotic initiation factor 4E gene in solid tumor. J Surg Res 85: 37-42. [Crossref]
- Zimmer SG, DeBenedetti A, Graff JR (2000) Translational control of malignancy: the mRNA cap-binding protein, eIF-4E, as a central regulator of tumor formation, growth, invasion and metastasis. Anticancer Res 20: 1343-1351. [Crossref]
- De Benedetti A, Graff JR (2004) eIF-4E expression and its role in malignancies and metastases. Oncogene 23: 3189-3199. [Crossref]
- Nakamura JL, Garcia E, Pieper RO (2008) S6K1 plays a key role in glial transformation. Cancer Res 68: 6516-6523. [Crossref]
- Bärlund M, Forozan F, Kononen J, Bubendorf L, Chen Y, et al. (2000) Detecting activation of ribosomal protein S6 kinase by complementary DNA and tissue microarray analysis. J Natl Cancer Inst 92: 1252-1259. [Crossref]
- Recher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, et al. (2005) Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 105: 2527-2534. [Crossref]
- Böhm A, Aichberger KJ, Mayerhofer M, Herrmann H, Florian S, et al. (2009) Targeting of mTOR is associated with decreased growth and decreased VEGF expression in acute myeloid leukaemia cells. Eur J Clin Invest 39: 395-405. [Crossref]
- Park S, Chapuis N, Saint Marcoux F, Recher C, Prebet T, et al. (2013) A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia 27, 1479-1486. [Crossref]
- Rizzieri DA, Feldman E, Dipersio JF, Gabrail N, Stock W, et al. (2008) A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 14: 2756-2762. [Crossref]
- Perl AE, Kasner MT, Tsai DE, Vogl DT, Loren AW, et al. (2009) A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in relapsed and refractory acute myelogenous leukemia. Clin Cancer Res 15: 6732-6739. [Crossref]
- Amadori S, Stasi R, Martelli AM, Venditti A, Meloni G, et al. (2012) Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: results of a phase II GIMEMA study (AML-1107). Br J Haematol 156: 205-212. [Crossref]
- Yee KW, Zeng Z, Konopleva M, Verstovsek S, Ravandi F, et al. (2006) Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 12: 5165-5173. [Crossref]
- Wei AH, Sadawarte S, Catalano J, Hills R, Avery S, et al. (2010) Phase Ib study combining the mTOR inhibitor everolimus (RAD001) with low-dose cytarabine in untreated elderly AML. Blood 116: 3299.
- Sehgal SN (2003) Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc 35: 7S-14S. [Crossref]
- Mita M, Sankhala K, Abdel-Karim I, Mita A, Giles F (2008) Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Exp Opin Invest Drugs 17: 1947-1954. [Crossref]
- Karen WL, Garcia-Manero G, Thomas D, Ravandi-Kashani F, Verstovsek S, et al. (2004) A phase II study of temsirolimus (CCI-779) in patients with advanced leukemias. Blood 104: 214b.
- Rizzieri DA, Feldman E, Moore JO, Roboz GJ, DiPersio JF, et al. (2005) A phase 2 clinical trial of AP23573, an mTOR inhibitor, in patients with relapsed or refractory hematologic malignancies. Blood 106: 835a.
- Xu Q, Thompson JE, Carroll M (2005) mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood 106: 4261-4268. [Crossref]
- Xu RH1, Pelicano H, Zhang H, Giles FJ, Keating MJ, et al. (2005) Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 19: 2153-2158. [Crossref]
- Nishioka C, Ikezoe T, Yang J, Koeffler HP, Yokoyama A (2008) Blockade of mTOR signaling potentiates the ability of histone deacetylase inhibitor to induce growth arrest and differentiation of acute myelogenous leukemia cells. Leukemia 22: 2159-2168. [Crossref]
- Park S, Chapuis N, Bardet V, Tamburini J, Gallay N, et al. (2008) PI-103, a dual inhibitor of class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia 22: 1698-1706. [Crossref]
- Papa V, Tazzari PL, Chiarini F, Cappellini A, Ricci F, et al. (2008) Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia 22: 147-160. [Crossref]
- Kojima K, Shimanuki M, Shikami M, Samudio IJ, Ruvolo V, et al. (2008) The dual PI3K/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53-wild type AML. Leukemia 22: 1728-1736. [Crossref]
- Fiedler W, Graeven U, Ergün S, Verago S, Kilic N, et al. (1997) Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood 89: 1870-1875. [Crossref]
- Aguayo A, Estey E, Kantarjian H, Mansouri T, Gidel C, et al. (1999) Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood 94: 3717-3721. [Crossref]
- Aguayo A, O'Brien S, Keating M, Manshouri T, Gidel C, et al. (2000) Clinical relevance of intracellular vascular endothelial growth factor levels in B-cell chronic lymphocytic leukemia. Blood 96: 768-770. [Crossref]
- Bellamy WT, Richter L, Sirjani D, Roxas C, Glinsmann-Gibson B, et al. (2001) Vascular endothelial cell growth factor is an autocrine promotor of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood 97: 1427-1434. [Crossref]
- Dias S, Hattori K, Zhu Z, Heissig B, Choy M, et al. (2000) Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest 106: 511-521. [Crossref]
- Dias S, Hattori K, Heissig B, Zhu Z, Wu Y, et al. (2001) Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc Natl Acad Sci U S A 98: 10857-10862. [Crossref]
- Böhm A, Aichberger KJ, Mayerhofer M, Herrmann H, Florian S, et al. (2009) Targeting of mTOR is associated with decreased growth and decreased VEGF expression in acute myeloid leukemia cells. Eur J Clin Invest 39: 395-405. [Crossref]
- Weisberg E, Banerji L, Wright RD, Barrett R, Ray A, et al. (2008) Potentiation of antileukemic therapies by the dual PI3K/PDK-1 inhibitor, BAG956: effects on BCR-ABL- and mutant FLT3-expressing cells. Blood 111: 3723-3734. [Crossref]
- Stauffer F, Maira SM, Furet P, García-Echeverría C (2008) Imidazo [4,5-c]quinolines as inhibitors of the PI3K/PKB-pathway. Bioorg Med Chem Lett 18: 1027-1030. [Crossref]
- Deng L, Jiang L, Lin XH, Tseng KF, Liu Y, et al. (2017) The PI3K/mTOR dual inhibitor BEZ235 suppresses proliferation and migration and reverses multidrug resistance in acute myeloid leukemia. Acta Pharmacol Sin 38: 382-391. [Crossref]
- Clemens MJ (2004) Targets and mechanisms for the regulation of translation in malignant transformation. Oncogene 23: 3180-3188. [Crossref]
- Kampa-Schittenhelm KM, Heinrich MC, Akmut F, Rasp KH, Illing B, et al. (2013) Cell cycle-dependent activity of the novel dual PI3K-mTORC1/2 inhibitor NVPBGT226 in acute leukemia. Mol Cancer 12: 46. [Crossref]
- Shah K, Moharram SA, Kazi JU (2018) Acute leukemia cells resistant to PI3K/mTOR inhibition display upregulation of P2RY14 expression. Clin Epigenetics 10: 83.
- Lee BC, Cheng T, Adams GB, Attar EC, Miura N, et al. (2003) P2Y-like receptor, GPR105 (P2Y14), identifies and mediates chemotaxis of bone marrow hematopoietic stem cells. Genes Dev 17: 1592-1604. [Crossref]
- Xu J, Morinaga H, Oh D, Li P, Chen A, et al. (2012) GRP105 ablation prevents inflammation and improves insulin sensitivity in mice with diet-induced obesity. J Immunol 189: 1992-1999.
- Sampath D, Malik A, Plunkett W, Nowak B, Williams B, et al. (2013) Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk Res 37: 1461-1467. [Crossref]
- Konopleva MY, Walter RB, Faderl SH, Jabbour EJ, Zeng Z, et al. (2014) Preclinical and early clinical evaluation of the oral AKT inhibitor, MK-2206, for the treatment of acute myelogenous leukemia. Clin Cancer Res 20: 2226-2235. [Crossref]
- Gojo I, Perl A, Luger S, Baer MR, Norsworthy KJ, et al. (2013) Phase I study of of UCN-01 and perifosine in patients with relapsed and refractory acute leukemias and high-risk myelodysplastic syndrome. Invest New Drugs 31: 1217-1227. [Crossref]
- Meja K, Stengel C, Sellar R, Huszar D, Davies BR, et al. (2014) PIM and AKT kinase inhibitors show synergistic cytotoxicity in acute myeloid leukaemia that is associated with convergence on mTOR and MCL1 pathways. Br J Haematol 2014, 167, 69-79. [Crossref]
- Rodrigues DA, Sagrillo FS, Fraga CAM (2019) Duvelisib: a 2018 novel FDA-approved small molecule inhibiting phosphoinositide 3-kinases. Pharmaceuticals (Basel) 12: 69. [Crossref]
- Pillinger, G.; Longhran, N.V.; Piddock, R.E. et al. (2016) Targeting PI3Kδ and PI3Kγ signaling disrupts human AML survivaland bone marrow stromal cell mediated protection. Oncotarget 7: 39784-39795. [Crossref]
- Yue EW, Li YL, Douty B, He C, Mei S, et al. (2019) INCB050465 (Parsaclisib), a novel next –generation inhibitior of phosphoinositide 3-kinase delta (PI3Kδ). ACS Med Chem Lett 10: 1554-1560. [Crossref]
- Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, et al. (2006) A selective inhibitor of the p110δ isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene 25: 6648-6659. [Crossref]
- Mirabilii S, Ricciardi MR, Piedimonte M, Gianfelici V, Bianchi MP, et al. (2015) Biological aspects of mTOR in leukemia. Int J Mol Sci 19: 2396. [Crossref]
- Rahmani M, Nkwocha J, Hawkins E, Pei X, Parker RE, et al. (2018) Cotargeting BCL-2 and PI3K induces BAX-dependent mitochondrial apoptosis in AML cells. Cancer Res 78: 3075-3086. [Crossref]
- Rahmani M, Aust MM, Hawkins E, Parker RE, Ross M, et al. (2015) Co-administration of the mTORC1/TORC2 inhibitor INK128 and the Bcl-2/Bcl-xL antagonist ABT-737kills humanmyeloid leukemia cells through Mcl-1 down-regulation and AKT inactivation. Haematologica 100: 1553-1563. [Crossref]
- Rahmani M, Aust MM, Atkisson E, Williams Jr DC, Ferreira-Gonzalez A, et al. (2013) Dual inhibition of Bcl-2 and Bcl-xL strikingly enhanced PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res 73: 1340-1351. [Crossref]
- Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D (2013) MEK and the inhibitors: from bench to bedside. J Hematol Oncol 6: 27. [Crossref]
- Nepstad I, Hatfield KJ, Gronningsæter IS, Reikvam H (2020) The PI3K-Akt-mTOR signaling pathway in human acute myeloid leukemia (AML) cells. Int J Mol Sci 21: 2907.
Editorial Information
Editor-in-Chief
Hiroshi Miyamoto
University of Rochester Medical Center
Article Type
Review Article
Publication history
Received: July 05, 2020
Accepted: July 25, 2020
Published: July 28, 2020
Copyright
©2020 Fuchs O. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
Fuchs O (2020) Targeting the phosphatidylinositol 3-kinase signaling pathway in acute myeloid leukemia. Integr Cancer Sci Therap 7: DOI: 10.15761/ICST.1000342.