TET enzymes are responsible for catalyzing the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), during the process of active DNA demethylation. These enzymes are differentially expressed in several tissues during development and can regulate several conserved signaling pathways, such as Wingless (WNT), Notch, Sonic Hedgehog (SHH) and Transforming Growth Factor Beta (TGF-β). Low expression of TET genes and the consequent reduction of 5hmC levels have been commonly reported in tumors of different origins and, in most cases, associated with poor prognosis. On this basis, we aimed to compile information about the canonical action of TET enzymes on the above signaling pathways during development, as well as the alterations characterized in different cancer cells. The presence of TETs is fundamental for normal embryonic development and their deletion in animal models has shown to delay cell differentiation and result in dysregulated expression of genes involved in signaling pathways. Consequently, the absence of TETs results in central nervous system defects and retinal deformity. In cancer, low expression of TETs induces activation of the WNT, TGF-β and NOTCH pathways, either directly or indirectly. Depletion in Tet activity inhibits tumorigenic processes, such as cell proliferation and epithelial-mesenchymal transition (EMT). The prospect of TET pharmacological or molecular manipulation might have global effects that should be considered for future therapeutic intervention.
TET enzymes, 5-hydroxymethylcytosine, signaling pathways, embryonic development, cancer
TET: Ten-Eleven Translocation (Human), Tet: Ten-eleven translocation (mouse), 5mC: 5-mehtylcytosine, 5hmC: 5-hydroxymethylcytosine, WNT: Wingless, SHH: Sonic Hedgehog, TGF-β: Transforming Growth Factor Beta, EMT: epithelial-mesenchymal transition, DSHB: double-stranded β-helix domain, IDAX: Dvl-binding protein, TDG: thymine DNA glycosylase, BER: base excision repair, H3K4me3: Tri methylation of lysine 4 in the histone 3, CNS: central nervous system, DC: destruction complex, APC: Apoptosis Poliposis Coli, GSK3β: Glycogen synthase kinase-3 Beta, CK1α/γ: casein kinase alfa/gama, CKα/o: casein kinase alfa/omega, FZD: Frizzled, LRP5/6: lipoprotein receptor-related protein5/6, TCF/LEF: T-cell factor/lymphoid enhancer factor, CRC: colorectal cancer, CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats), C-myc: cancer – Myelocytomatosis, Sfrp: secreted frizzled-related protein , DKK: Dickkopf-related protein, HH: Hedgehog, IHH: Indian Hedgehog, DHH: Desert Hedgehog, PTCH1: Patched-1, SMO: Smothened, GLI: glioma-associated oncogene, SUFU: suppressor of Fused, BCL2: B-cell lymphoma 2, ANG1/2: angiopoietin, SNAIL: Zinc finger protein SNAI1, Sox2: SRY-box 2, ChIP: chromatin immunoprecipitation, Hhip: hedgehog interacting protein, Pax1/9: Paired box 1, Ccnd2: Cyclin D2, BMPs: bone morphogenetic proteins, GDF: growth and differentiation factors, SMAD: SMAD family member, FOXH1: Forkhead box protein H1, MPKs: mitogen-activated protein kinase, CDKs: cyclin dependent kinases, Smurf: SMAD ubiquitination regulatory factor, DNMT: DNA methyltransferase, PEN2: presenilin enhancer 2, APH1: Homolog B, Gamma-Secretase Subunit, NICD: Notch intracellular domain, RBP-J: recombination signal sequence-binding protein J, Hes-1: Hes Family BHLH Transcription Factor 1, Jag (1,2): Jagged
DLL (1/3): Delta-like, HES5: Hes Family BHLH Transcription Factor 5, DNER: Delta/Notch Like EGF Repeat Containing, GFAP: Glial Fibrillary Acidic Protein, AXL1: AXL receptor tyrosine kinase
Hydroxymethylation is an epigenetic mechanism with a crucial role during development of mammals. 5-hydroxymethylcytosine (5hmC) influences chromatin structure and genomic function [1]. High levels of 5hmC are present in regulatory regions and along expressed genes and are associated with the recruitment of the transcriptional machinery. Conversion of 5mC to 5hmC is catalyzed by TET (Ten Eleven Translocation) enzymes: TET1, TET2 and TET3, which constitute a family of 2-oxoglutarate-dependent dioxygenase and iron (II). These proteins display a catalytic region in their C-terminal, with methylcytosine dioxygenase activity, composed by a conserved cysteine (Cys-rich) rich domain; and a double-stranded β-helix domain (DSHB), which interacts with 2-oxoglutarate and iron (II). At the N-terminal region, TET1 and TET3 show a CXXC domain, which binds to unmodified cytosines [1]. Conversely, as a consequence of a chromosome inversion during evolution, this domain is not present in TET2; the CXXC was separated and gave origin to the IDAX gene, which acts as its negative regulator (Figure 1) [2].
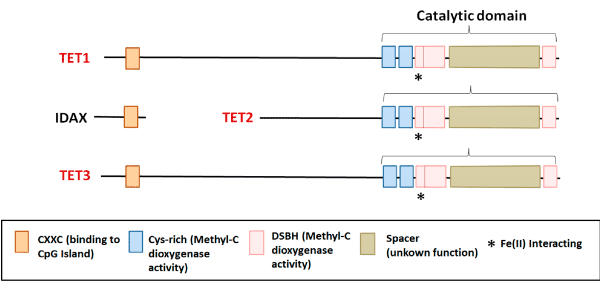
Figure 1. TETs showed a catalytic region in their C-terminal, with methylcytosine dioxygenase activity, composed by a conserved domain rich in cysteine (Cys-rich); and a double-stranded β-helix domain (DSHB), which interacts with 2OG and Fe (II). At the N-terminal region, TET1 and TET3 show a CXXC domain, which binds to unmodified cytosine; Conversely, as a consequence of a chromosome inversion during evolution, this domain is not present in TET2; the CXXC was separated and gave origin to the IDAX gene, which acts as its negative regulator.
During the process of active DNA demethylation, TET enzymes oxidize the methyl group to 5hmC, 5-formylcytosine and 5-carboxylcytosine (Figure 2). The bases are then recognized and excised by the enzyme thymine DNA glycosylase (TDG) and substituted by an unmodified cytosine by base excision repair (BER) [3-5]. Independent of their catalytic activity, TET2 and TET3 are also associated with chromatin remodeling and H3K4me3 mark (Tri methylation of lysine 4 in histone 3), a histone mark indicative of transcriptionally permissive chromatin states [6].
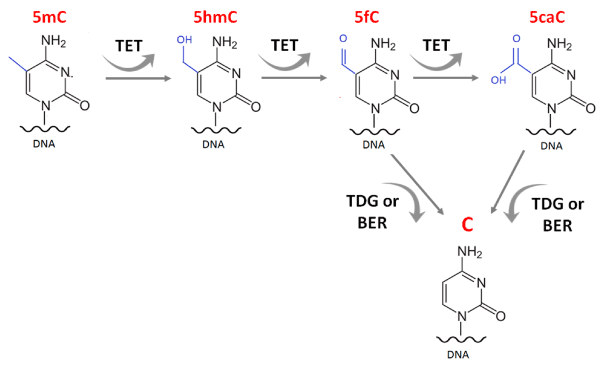
Figure 2. TET enzymes in the process of DNA demethylation. TET1/2/3 oxidize the methyl group into 5hmC, 5-formylcytosine and 5-carboxylcytosine. These bases are recognized and excised by the enzyme thymine DNA glycosylase (TDG) and substituted by an unmodified cytosine by base excision repair pathway (BER).
During the initial phases of embryonic development, TET enzymes are differentially expressed in several tissues, and have a specific expression profiles for each cell type, since the beginning of gastrulation [7-11]. Expression levels of TET enzymes (TET1 and TET2) in embryonic stem cells of animal models showed that these enzymes play important roles in tissue control and maintenance at embryonic level [12,13]. During central nervous system (CNS) development, these enzymes take part in several important biological processes, influencing oligodendrocyte maturation and differentiation, and myelination of cells [14].
TET1 is highly expressed in early blastocyst cells, and it is crucial in regulating the proliferation of neural progenitor cells [15]. Therefore, changes in its function can promote phenotype anomalies at the hippocampus and decreasing neurogenesis by transcription repression [15]. TET2 has been identified as responsible for maintaining hematopoietic stem cells and establishing DNA hypomethylation during neuronal tissues development. Likewise, TET3 is essential for the maintenance of neuronal progenitor cells in neocortical development [12,16-18].
Furthermore, TET1 and TET2 perform important functions during postnatal development. TET1 forms repressive complexes binding to target gene promoters, assuming an important role in transcriptional activation of tissues in development; [13] while the absence of TET2 may impair the maintenance of hematopoietic stem cells contributing to the development of acute myelomonocytic leukemia [12,19]. Therefore, TET enzymes activities are important in several stages of cell differentiation in tissue development, and any dysregulation, can lead to complications and loss of specific functions in each cellular context [20-22].
In cancer, decreased expression of TET genes and the consequent reduction of 5hmC levels have been frequently reported in tumors of different origins, and related to a poor prognosis [23-31]. Low 5hmC and 5mC levels are associated with the accumulation of methylation in specific promoter regions of tumor suppressor genes related to cancer progression [25,32,33]. Moreover, decreased 5hmC levels are associated with increased genomic instability [34].
A recent study demonstrated a similarity between the DNA methylation patterns of TET-deficient cells types and the profile of cancer cells, suggesting that local hypermethylation in euchromatin and DNA hypomethylation in heterochromatin can be associated with oncogenic transformation [35].
Manipulation of TET expression during embryonic development and in cancer cell lines has demonstrated the relevance of these genes in the control of conserved signaling pathways, such as Wingless (WNT), Notch, Sonic Hedgehog (SHH) and Transforming Growth Factor Beta (TGF-β), whose dysregulation is generally associated with tumor progression. In this review, we propose to analyze the action of TET enzymes in these signaling pathways in development, as well as the alterations displayed in cancer cells.
TETs and WNT pathway
The WNT canonical signaling causes the accumulation of β-catenin in the cytoplasm and its eventual translocation into the nucleus to act as a transcriptional coactivator of transcription factors. When inactive, β-Catenin stability is regulated by a destruction complex (DC) [36,37]. Consisting of AXIN, the tumor suppressor Adenomatous Polyposis Coli (APC) and two activated kinases, the GSK3β (Glycogen Synthase Kinase 3 Beta) and CK1α/γ (Casein Kinase 1 Alpha/Gama). This complex, leads to the phosphorylation of β-Catenin, by CK1α/γ and GSK3β, and consequent proteasomal degradation. WNT ligands binding to receptor co-operators of Frizzled (FZD) and the lipoprotein receptor-related protein5/6 (LRP5/6) [38] and leads to their dimerization and the phosphorylation of the intracellular tail of LRP5/6 by GSK3β at serine 9. Consequently, the DC is abrogated and β-Catenin accumulates in the cytoplasm and nucleus where it binds to TCF/LEF (T-cell factor/lymphoid enhancer factor) and activates the transcription of WNT target genes: AXIN2 and leucine rich repeat containing G protein-coupled receptor 5 (LGR5), the key effectors of WNT signaling [39,40].
In tissue development, this pathway is constitutively activated until it reaches the precise stage and differentiation [41]. However, inactivating events on the DC or mutations in phosphorylation coding sites of genes belonging to WNT core components, lead to several pathologies, including cancer [41]. In recent years, WNT pathway components have frequently been described as over- or underexpressed in different human cancers playing key roles in tumor initiation, tumor growth, cell senescence, cell death, and metastasis [42]. In medulloblastomas, for example, the WNT canonical pathway is often activated and patients are categorized within the WNT molecular subgroup; and are assigned with a good prognosis [43]. Likewise, in breast and colon carcinomas, high levels of nuclear β-Catenin are usually considered predictors of increased WNT signaling activity and correlate with poor prognosis [44,45].
Alternatively, loss of APC is a driver event in colorectal cancer (CRC) and is associated with poor prognosis. Notably, tumor phenotype is reversed to normal as demonstrated by APC knockout in vivo models of CRC [46,47]. In other tumors, i.e. melanoma, the WNT pathway seems delays senescence [48], and mutations that disrupt the phosphorylation and degradation of β-Catenin are frequent in hepatocellular carcinoma and ovarian cancer, whereas mutations in AXIN1 are common in colorectal tumors, most of which serve as prognostic indicators [49,50].
The investigation of Tet1 deletions in mice also showed that this gene is required to maintain the morphology of epithelial cells and regulate the WNT pathway. Tet1 absence resulted in increased 5mC levels and decreased 5hmC levels at the promoter regions of Axin2, C-Myc and Sox-9 genes in intestinal stem cells isolated from mice [51]. Tet3 also displayed key roles in the regulation of the WNT pathway, controlling the balance between mesodermal and neuroectodermal fate in mouse embryonic stem cells on the generation of neural structures [52]. In mouse embryos, Tet3 demethylates the promoter region and activates the expression of secreted frizzled-related protein 4 (Sfrp4) gene, which is responsible for inhibiting the WNT pathway [52]. Moreover, the combination of transcriptome techniques and pharmacological manipulation, proved that Tet2 and Tet3 regulate the Notch and WNT pathways in zebrafish [53], and their inactivation during retinal neurogenesis results in the absence of a clear “zone” of differentiation and upregulation of Wnt9b [53].
Alternatively, in human cancer, the action of TET1 enzyme was demonstrated upstream the WNT pathway, and regulate DKK (Dickkopf) and SFRP genes expression by demethylation, both repressor genes of this pathway. In colon cancer, TET1 gene knockout increases proliferation, while its induction, inhibits the proliferation and growth of xenograft tumors [54]. TET1 maintains the inhibitors of the pathway hypomethylated; its upregulation induces the activation of DKK3 and DKK4, and the downregulation of Myc and CyclinD2 [54]. Moreover, chromatin immunoprecipitation assays demonstrated the TET1 binding at the promoters of DKK genes, as well as increased levels of 5hmC and the concomitant decrease in 5mC [54]. In epithelial ovarian cancer, the expression of TET1 was inversely correlated with the clinical advance of the disease; while its upregulation inhibited colony forming, cell migration and invasion in SKOV3 and OVCAR3 cell lines, and EMT in SKOV3. Additionally, TET1 inhibited the WNT/β-Catenin pathway (or canonical WNT pathway), through the demethylation and upregulation of sFRP2 and DKK1 (Figure 3A) [55].
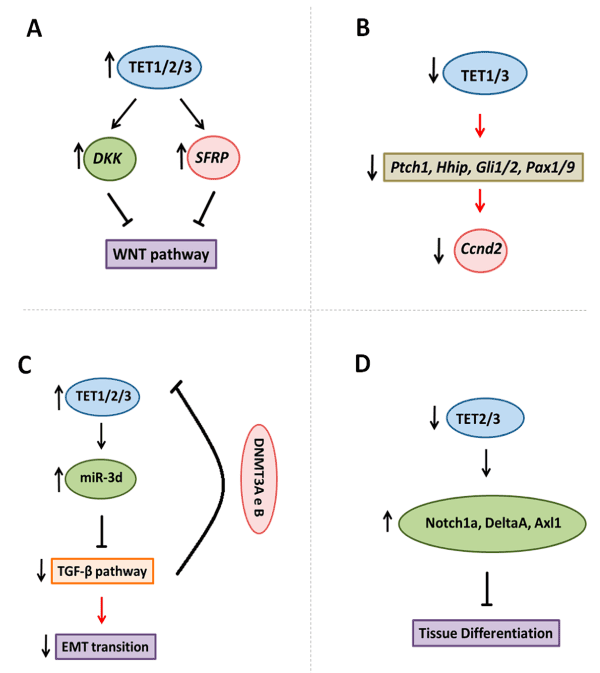
Figure 3. TET enzymes target signalling pathways. A. TET enzymes demethylated DKK and SFPR genes that inhibited WNT pathway in embryonic development and cancer. B. Tet1/3 knockout is associated with decreased Ptch1, Hhip, Gli1/2 and Pax1/9 genes, that are members of SHH pathway, and reduced downstream genes such as Ccnd2. C. TET3 demethylates the precursor of miR-3d, and thus, inhibiting TGF-β pathway, and reducing epithelia-mesenchymal transition. Whereas TGF-β pathways stimulated DNMT3A and B activity in promoter of TET2 and 3, with a reduced in these genes expression. D. The control of NOTCH signaling components by Tet2 e Tet3 enzymes.
Tumor progression in melanoma was also associated with changes in 5hmC levels, and the enrichment of pathways by the Kyoto Encyclopedia of Genes and Genome evidenced the accumulation of 5mC in genes associated with the WNT pathway [25]. In medulloblastoma, the subgroup that displays activation of the WNT pathway showed lower expression of TET1, pointing out a possible association between its downregulation and the activation of the pathway [56].
TETs and SHH pathway
The canonical Hedgehog (HH) pathway consists, mainly, of the glycoproteins Sonic Hedgehog (SHH), Indian Hedgehog (IHH) and Desert Hedgehog (DHH) [57]. When SHH is secreted, it binds to the 12-transmembrane protein Patched-1 (PTCH1), a Smothened (SMO) inhibitor. SHH binding promotes abrogation of Patched-1 activity, releasing SMO. This event leads to the nuclear localization of glioma-associated transcription factors (GLI1, GLI2 and GLI3), considered the key effectors of this pathway [58].
Another important component of the SHH pathway, and repressor of GLI, is the suppressor of Fused (SUFU). SUFU is a negative regulator of the SHH pathway that directly binds to GLI, causing its arrestment, processing or degradation in the cytoplasm [59]. This mechanism prevents activation of SHH pathway target genes [60]. GLI activity can be involved in the activation of several genes including CCND1 (Cyclin-D1, cell cycle), MYC (Proliferation), BCL2 (Apoptosis), ANG1/2 (Angiogenesis), SNAIL (EMT), NANOG and SOX2 (Stem cells self-renewal); and in the positive feedback loop stimulating transcription of GLIs and PTCH1 [61-65].
In cancer, the SHH pathway is aberrantly activated due to mutations in PTCH1, SUFU or SMO. Persistent activation of the pathway stabilizes the expression of GLI1 and GLI2 that promotes proliferation and invasion and inhibits apoptosis [58].
Specifically in medulloblastoma, the SHH pathway plays an important role in the molecular subgroup assigned as SHH-medulloblastoma (SHH-MB) [58]. In rhabdomyosarcoma, the most frequent soft tissue sarcoma in children, which is divided into two major histological subgroups, embryonal and alveolar, is described to show HH/SMO pathway activation [65]. Embryonal Tumors are characterized by SMO localization in primary cilia and/or GLIs hyperexpression. These features are almost exclusively found in tumors with activated SHH signaling pathway [43,65]. Clinical trials are being conducted to test SMO inhibitors, such as Vismodegib or Sonidegib. Nonetheless, despite initial favourable outcomes, some patients still present resistance or relapse [43].
SHH is also controlled by TET enzymes. Xu et al. [66] demonstrated the role of Tet3 5mC hydroxylase and its CXXC domain in upstream transcriptional regulation of genes important in early eye and neural development, through loss-of-function studies focused on Tet3 and using Xenopus sp. as an experimental model. Depletion of Tet3 exhibited effects in a set of key development genes, including, two major SHH signaling components: shh and ptc-1, that showed low expression in embryos at stage 14, in the absence of Tet3 [66]. Moreover, chromatin immunoprecipitation (ChIP) assay demonstrated the binding of Tet3 enzyme in promoter region of shh and ptc-1 [66] (Figure 3B). In parallel, regulation of the 5hmC status at its targets showed a significant reduction in 5hmC at CCGG sites in ptc-2 gene promoter, after Tet3 depletion [66]. Of note, the CXXC domain of TET3 has DNA interaction and binding to restricted genomic regions, including the PTCH1 and SHH genes in HEK293T cells [66].
The control of SHH by Tets can also occur indirectly. Tet1/Tet3 knockout, in mice (Tet1/3 DKO embryos), induced defects in cholesterol synthesis; since SHH signaling requires covalent modification of cholesterol for its activity during development. Significant decrease in expression of SHH components, such as Ptch1, hedgehog interacting protein (Hhip), transcription factors (Gli1/2, Pax1/9) and other pathway targets, such as Cyclin D2 (Ccnd2) was also observed in this model (Figure 3B). Dysregulation in SHH function contributes to developmental abnormalities [67]. Late stage Tet1/3 DKO exhibited poor forebrain formation and abnormal facial structures, with phenotypes observed in the dysfunction of SHH [66]. Thus, the function of TETs enzymes is fundamental for the SHH pathway activation during development. In cancer, medulloblastoma cell lines classified as SHH subgroup showed high levels of TET1 expression [68]. However, there are no functional descriptions of TET and SHH or about any correlation between them in other tumor types.
TETs and TGF-β
The transforming growth factor-β (TGF-β) family consists of three TGF-β isoforms (TGFB1, TGFB2 and TGFB3), NODAL, bone morphogenetic proteins (BMPs), and growth and differentiation factors (GDFs) [69]. These proteins share features regarding signaling, regulation and structure, however, the specific function and complexity of the canonical and non-canonical pathways remain to be elucidated [69-72].
The activation of this pathway requires the ligand binding to one of the five membranous type II serine/threonine kinase receptors and recruitment and transphosphorylation of one of the seven type I serine/threonine kinase receptors [69]. After activation by phosphorylation, the type I receptor kinase domain triggers the signal of the canonical pathway, which comprises the phosphorylation of the SMAD-dependent axis, including receptor-regulated SMAD proteins (R-SMAD) [69]. During the canonical signaling, SMAD2 and SMAD3 with NODAL, activin and TGFB are consistently correlated. Alternatively, some authors also describe that TGFB possibly induces phosphorylation of SMAD1 and SMAD5, as alternative signals triggered by BMPs and GDFs [70,72]. During downstream signaling, SMAD2 and SMAD3 form complexes and translocate into the nucleus after phosphorylation. In the nucleus, after accumulation and binding to cofactors, such as p300-CBP and FOXH1, they trigger transcription of several target genes [73].
Other potential regulators of TGF-β pathway are the Mitogen Activated Protein Kinases (MAPKs), Glycogen synthase kinase 3β (GSK3β), cyclin dependent kinases (CDKs) and Smurf (SMAD ubiquitination regulatory factor). Altogether, they can modulate SMADs activities in different manners, including phosphorylation and ubiquitination [69].
The TGFB family components are involved in early embryonic development and tissue homeostasis in adults. Nonetheless, their roles in cellular growth, differentiation, apoptosis, extracellular matrix production, EMT and immune-response, have only been partially elucidated [69]. However, when aberrantly activated or inactivated, depending on the cellular context, they might promote different pathologies [74].
Regarding carcinogenesis and tumor initiation, the TGF-β pathway shows duality: in the early stages, it might act as a tumor suppressor, though later, it might stimulate a pro-carcinogenesis microenvironment, promoting in most cases, metastasis [74]. Several studies have demonstrated that TGFBR1, TGFBR2, SMAD2 and SMAD4 are frequently inactivated through mutation, allelic heterozygosity loss or inactivation through methylation [70]. In SHH medulloblastoma, high SMAD2 expression and IHC staining in patients samples was associated with good prognosis while, in Group 3 medulloblastoma, the SNAIL/TGF-β axis is associated with metastasis and promotes EMT [75]. In others tumors (i.e., melanoma, renal cell carcinoma, mesothelioma, glioma and pancreatic ductal adenocarcinoma) there are several ongoing clinical trials utilizing TGF-β inhibitors to target TGFB1 or TGFB2 or both [74]. Fresolimumab, a TGF-β inhibitor for TGFB1, B2 and B3 and PF-03446962(®Pfizer) an ALK inhibitor, are the most widely used. Nonetheless, despite the initial good outcomes, the authors suggested the combination of TGF-β inhibitors with the current standard chemotherapy or epigenetic modulators to improve the efficiency of multimodal therapy [74].
In ovarian cancer, TGFB1 induced EMT with downregulation of miR-30 family and the restoration of miR-30d inhibited this phenotype. Moreover, the action of TET3 in TGF-β pathway in this model, occurred through the demethylation of miR-30d precursor [76]. Furthermore, when ovarian cancer cell lines SKOV3 and 3OA were treated with TGF-β1 (10ng/mL), they showed downregulation of TET1 and TET3, although, no alteration in TET2 was observed. TET3 was the most significantly reduced and when upregulated inhibited the TGF-β pathway and resulted in the blockade of EMT with the concomitant reduction of the EMT markers, E-cadherin, Vimentin, N-cadherin and Snail. Therefore, cells with TET3 expression exhibited decreased invasive and migratory capabilities [76]. Also, it has been suggested that the TGF-β pathway could regulate TET2 and TET3 genes expression, by stimulating the expression and recruitment of DNMT (DNA methyltransferase) 3A and 3B to their promoter regions, inducing hypermethylation and consequent silencing of these genes (Figure 3C) [77].
TETs and notch pathway
The Notch signalling pathway is critical for embryonic development. This pathway is activated by interactions between adjacent cells in order to contribute to the formation, growth, and development of embryos [78]. Even though Notch was first classified as a neurogenic gene, the characterization of Drosophila embryos made it clear that this pathway is highly pleiotropic [79]. Notch signaling has been described to be activated in somite formation (somitogenesis), heart formation (cardiogenesis), muscle tissue formation (myogenesis), hematopoiesis, vasculogenesis and angiogenesis, among others [78,80,81].
In mammals, there are four Notch receptors, NOTCH 1-4, and five transmembrane ligands, Jagged1 (JAG1), Jagged2 (JAG2), Delta-like1 (DLL1), Delta-like3 (DLL3), and Delta-like4 (DLL4), which interact with each other to activate the pathway. At the molecular level, the binding between Notch receptors and their ligands promotes two proteolytic cleavage events at the receptors. The first cleavage is catalysed by the ADAM-family of metalloproteases and the second cleavage is mediated by ɣ-secretase, an enzyme complex that contains PRESENILIN, NICASTRIN, PEN2 and APH1. These cleavages release the Notch intracellular domain (NICD), which translocates into the nucleus and acts as a transcriptional coactivator. NICD cannot bind directly to DNA but heterodimerizes with the DNA-binding protein RBP-J (recombination signal binding protein for immunoglobulin kappa J)/CBF1, forming the CSL complex (CBF1, Su(H) and LAG-1), thus, activating the transcription of Notch-target genes [82].
Notch pathway was described for the first time in leukemia, however, is also involved in other tumors. In breast cancer, low expression of NUMB, a negative regulator of Notch pathway, and an increased NOTCH1 expression were described in approximately 50% of primary human mammary carcinomas [83]. Likewise, in medulloblastoma, higher expression of NOTCH2 was shown in 15% of the cases and high expression of HES1, a target of Notch signaling, was associated with decreased survival [84].
TETs enzymes and 5hmC, also play relevant roles in controlling the expression of molecules involved in the Notch signaling pathway. Some studies have demonstrated a relationship between Notch signaling genes and epigenetic modifications in biological processes. Notch pathway plays a critical role in osteoblast cell fate and function, and endothelial Notch activity is associated with stimulation of angiogenesis and osteogenesis [85]. It was reported that TET3 and 5hmC were upregulated in dexamethasone-treated MLO-Y4 osteocyte-like cells and pathways enrichment analysis by KEGG showed that Notch signaling pathway exhibited dynamic changes in 5hmC levels and showed the most significant 5hmC downregulation, among the other signaling pathways, in those cells [86]. Changes in NOTCH4 expression were also found in steroid-associated osteonecrosis tissue suggesting that Notch signaling may also partially mediate the functional effect of 5hmC changes [86].
During differentiation of embryonic midbrain-type neural precursor cells derived from human embryonic stem cells occurred an increase of NOTCH1 expression, with gains in 5hmC at the gene body and a loses of 5mC at the promoter and gene body. Besides that, the NOTCH1 signaling pathway target genes DLL1, HES5, DNER and GFAP also gained 5hmC [87]. Moreover, significant hypomethylation was observed using bisulfite sequencing in myoblasts, myotubes, and skeletal muscle when compared to non-muscle samples at intragenic or intergenic regions of the following Notch receptor or ligand genes: NOTCH1, NOTCH2, JAG2, and DLL1 [88]. In parallel, an enzymatic assay of sites within or near these genes revealed an unusually high enrichment of 5hmC (up to 81%) in skeletal muscle, heart, and cerebellum [88]. Moreover, a study from Li, C et al. [89] demonstrated that Notch signaling in the hemogenic endothelium is regulated by Tet2/3 [89], while, it was [53] showed that occur the control of Notch signaling by Tet2 e Tet3 during zebrafish neurogenesis. In the absence of Tets, there is an upregulation of Notch1a, DeltaA, Axl1 genes (Figure 3D); and in Tet2 and Tet3 mutants the retina did not differentiate properly [53].
In this review, we discussed the role of TETs enzymes and their association with signaling pathways in embryonic development and cancer. The expression of TETs is fundamental for normal embryonic development and their deletion in animal models has shown to delay cell differentiation and result in dysregulated expression of genes involved in signaling pathways. Consequently, the absence of TETs results in central nervous system defects and retinal deformity. In cancer, low expression of TETs induces activation of the WNT, TGF-β and NOTCH pathways, either directly or indirectly. Depletion in Tet activity inhibits tumorigenic processes, such as cell proliferation and epithelial-mesenchymal transition (EMT). The prospect of TET pharmacological or molecular manipulation might have global effects that should be considered for future therapeutic intervention.
Declarations
Availability of data and material: Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Competing interests: The authors declare that they have no competing interests.
Funding: This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, process number 2018/05401-7 and 2014/20341-0).
Authors’ contributions: KBS wrote, organized the manuscript, and created the figures. GAVG, PFC and RB wrote the manuscript. MSB and LGT edited and finalized the manuscript. All authors have read and approved the final manuscript. The authors declare that this article is original, has never been published, and has not been submitted to any other journal.
Authors’ information: KBS, GAVG and RB are Postdoctoral fellow at Pediatric Oncology Laboratory, at the Department of Pediatrics, Ribeirao Preto Medical School, University of Sao Paulo, Brazil. PFC is PhD student in the Department of Genetics, Ribeirao Preto Medical School, University of Sao Paulo, Brazil. MSB is Professor at the Department of Biology from the Faculty of Philosophy, Sciences and Letters at Ribeirao Preto, Brazil. LGT is Professor in the Department of Pediatrics, Ribeirao Preto Medical School, University of Sao Paulo, Brazil.
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1.Science324: 930-935. [Crossref]
- Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, et al. (2015) TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev 263: 6-21. [Crossref]
- An J, Rao A, Ko M (2017) TET family dioxygenases and DNA demethylation in stem cells and cancers.Exp Mol Med49: e323. [Crossref]
- Cortazar D, et al. (2011) Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470: 419. [Crossref]
- Kohli RM, Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation.Nature502: 472-479. [Crossref]
- Mariappa D, Pathak S, van Aalten DM (2013) A sweet TET-à-tête-synergy of TET proteins and O-GlcNAc transferase in transcription.EMBO J32: 612-613. [Crossref]
- Colquitt BM, Allen WE, Barnea G, Lomvardas S (2013) Alteration of genic 5-hydroxymethylcytosine patterning in olfactory neurons correlates with changes in gene expression and cell identity. Proc Natl Acad Sci U S A 110: 14682-14687. [Crossref]
- Song SJ, Poliseno L, Song MS, Ala U, Webster K, et al. (2013) MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 154: 311-324. [Crossref]
- Dai HQ, Wang BA, Yang L, Chen JJ, Zhu GC, et al. (2016) TET-mediated DNA demethylation controls gastrulation by regulating Lefty–Nodal signaling. Nature 538: 528-532. [Crossref]
- Scott-Browne JP, Lio CJ, Rao A (2017) TET proteins in natural and induced differentiation.Curr Opin Genet Dev46: 202-208. [Crossref]
- Melamed P, Yosefzon Y, David C, Tsukerman A, Pnueli L (2018) Tet Enzymes, Variants, and Differential Effects on Function. Front Cell Dev Biol 6: 22. [Crossref]
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, et al. (2011) Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8: 200-213. [Crossref]
- Wu H, D'Alessio AC, Ito S, Xia K, Wang Z, et al. (2011) Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 473: 389. [Crossref]
- Zhao X, Dai J, Ma Y, Mi Y, Cui D, et al. (2014) Dynamics of ten-eleven translocation hydroxylase family proteins and 5-hydroxymethylcytosine in oligodendrocyte differentiation. Glia 62: 914-926. [Crossref]
- Zhang R, Cui QY, Murai K, Lim YC, Smith ZD, et al. (2013) Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. 13: 237-245. [Crossref]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, et al. (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell 20: 11-24. [Crossref]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA,et al. (2013) Global epigenomic reconfiguration during mammalian brain development. Science 341: 1237905. [Crossref]
- Lv X, Jiang H, Liu Y, Lei X, Jiao J (2014) MicroRNA-15b promotes neurogenesis and inhibits neural progenitor proliferation by directly repressing TET3 during early neocortical development. EMBO Rep 15: 1305-1314. [Crossref]
- Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, et al. (2013) Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell 24: 310-323. [Crossref]
- Chen E, Schneider RK, Breyfogle LJ, Rosen EA, Poveromo L, et al. (2015) Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood 125: 327-335. [Crossref]
- Cimmino L, Dawlaty MM, Ndiaye-Lobry D, Yap YS, Bakogianni S, et al. (2015) TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol 16: 653. [Crossref]
- Rasmussen KD, Helin K (2016) Role of TET enzymes in DNA methylation, development, and cancer.Genes Dev30: 733-750. [Crossref]
- Haffner MC, Chaux A, Meeker AK, Esopi DM, Gerber J, et al. (2011) Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2: 627-637. [Crossref]
- Kudo Y1, Tateishi K, Yamamoto K, Yamamoto S, Asaoka Y, et al. (2012) Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci 103: 670-676. [Crossref]
- Lian CG, Xu Y, Ceol C, Wu F, Larson A, et al. (2012) Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma.Cell150: 1135-1146. [Crossref]
- Liu C, Liu L, Chen X, Shen J, Shan J, et al. (2013) Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PloS One 8: e62828. [Crossref]
- Yang H, Liu Y, Bai F, Zhang JY, Ma SH, et al. (2013) Tumor development is associated with decreased of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 32: 663-669. [Crossref]
- Frycz BA, Murawa D, Borejsza-Wysocki M, Marciniak R, Murawa P, et al. (2014) Decreased expression of ten-eleven translocation 1 protein is associated with some clinicopathological features in gastric cancer. Biomed Pharmacother 68: 209-212. [Crossref]
- Dong ZR, Zhang C, Cai JB, Zhang PF, Shi GM, et al. (2014) Role of 5-hydroxymethylcytosine level in diagnosis and prognosis prediction of intrahepatic cholangiocarcinoma. Tumor Biol 4: 2763-2771. [Crossref]
- Feng J, Wang Q, Li G, Zeng X, Kuang S, et al. (2015) TET1 mediated diferente transcriptional regulation in prostate cancer. Int J Clin Exp Med 8: 203-211. [Crossref]
- Murata A, Baba Y, Ishimoto T, Miyake K, Kosumi K, et al. (2015) TET family proteins and 5-hydroxymethylcytosine in esophageal squamous cell carcinoma.Oncotarget6: 23372-23382. [Crossref]
- Uribe-Lewis S, Stark R, Carroll T, Dunning MJ, Bachman M, et al. (2015) 5-hydroxymethylcytosine marks promoters in colon that resist DNA hypermethylation in cancer. Genome Biol 16: 69-84. [Crossref]
- Williams K, Christensen J, Helin K (2011) DNA methylation: TET proteins-guardians of CpG islands?EMBO Rep13: 28-35. [Crossref]
- Tomkova M, McClellan M, Kriaucionis S, Schuster-Boeckler B (2016) 5-hydroxymethylcytosine marks regions with reduced mutation frequency in human DNA. ELife. [Crossref]
- López-Moyado IF, Tsagaratou A, Yuita H, Seo H, Delatte B, et al. (2019) Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc Natl Acad Sci U S A 116: 16933-16942. [Crossref]
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) beta-catenin is a target for the ubiquitin-proteasome pathway.EMBO J16: 3797-3804. [Crossref]
- Kitagawa M1, Hatakeyama S, Shirane M, Matsumoto M, Ishida N, et al. (1999) An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J 18: 2401-2410. [Crossref]
- Stamos JL, Chu ML, Enos MD, Shah N, Weis WI (2014) Structural basis of GSK-3 inhibition by N-terminal phosphorylation and by the Wnt receptor LRP6.Elife3: e01998. [Crossref]
- Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, et al. (2012) Wnt signaling through inhibition of β- catenin degradation in an intact Axin1 complex.Cell149: 1245-1256. [Crossref]
- Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, et al. (2014) YAP/TAZ incorporation in the b-catenin destruction complex orchestrates the Wnt response. Cell 158: 157-170. [Crossref]
- Losick VP, Morris LX, Fox DT, Spradling A (2011) Drosophila stem cell niches: a decade of discovery suggests a unified view of stem cell regulation.Dev Cell21: 159-171. [Crossref]
- Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer.Nat Rev Cancer13: 11-26. [Crossref]
- Wang J, Garancher A, Ramaswamy V, Wechsler-Reya RJ (2018) Medulloblastoma: From Molecular Subgroups to Molecular Targeted Therapies.Annu Rev Neurosci41: 207-232. [Crossref]
- López-Knowles E, Zardawi SJ, McNeil CM, Millar EK, Crea P, et al. (2010) Cytoplasmic localization of beta-catenin is a marker of poor outcome in breast cancer patients. Cancer Epidemiol Biomarkers Prev 19: 301-309. [Crossref]
- Chung GG, Provost E, Kielhorn EP, Charette LA, Smith BL, et al. (2001) Tissue microarray analysis of beta-catenin in colorectal cancer shows nuclear phospho-beta-catenin is associated with a better prognosis. Clin Cancer Res 7: 4013-4020 [Crossref]
- Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, et al. (2015) Sequential cancer mutations in cultured human intestinal stem cells. Nature 521: 43-47. [Crossref]
- Matano M, Date S, Shimokawa M, Takano A, Fujii M,et al. (2015) Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med 21: 256-262. [Crossref]
- KAUR, Amanpreet; WEBSTER, Marie R; WEERARATNA, Ashani T. () In the Wnt-er of life: Wnt signalling in melanoma and ageing. Br J Cancer 115: 1273-1279. [Crossref]
- Salahshor S, Woodgett JR (2005) The links between axin and carcinogenesis.J Clin Pathol58: 225-236. [Crossref]
- Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, et al. (2000) AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 24: 245-250. [Crossref]
- Kim R, Sheaffer KL, Choi I, Won KJ, Kaestner KH (2016) Epigenetic regulation of intestinal stem cells by Tet1-mediated DNA hydroxymethylation.Genes Dev30: 2433-2442. [Crossref]
- Li X, Yue X, Pastor WA, Lin L, Georges R, et al. (2016) Tet proteins influence the balance between neuroectodermal and mesodermal fate choice by inhibiting Wnt signaling. Proc Natl Acad Sci U S A 113: E8267-E8276. [Crossref]
- Seritrakul P, Gross JM (2017) Tet-mediated DNA hydroxymethylation regulates retinal neurogenesis by modulating cell-extrinsic signaling pathways. PLoS Genet 13: e1006987. [Crossref]
- Neri F, Dettori D, Incarnato D, Krepelova A, Rapelli S, et al. (2015) TET1 is a tumor suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 34: 4168-4176. [Crossref]
- Duan H, Yan Z, Chen W, Wu Y, Han J, et al. (2017) TET1 inhibits EMT of ovarian cancer cells through activating Wnt/ß-catenin signaling inhibitors DKK1 and SFRP2. Gynecol Oncol 147: 408-417. [Crossref]
- Bezerra Salomão K, Cruzeiro GAV, Bonfim-Silva R, Geron L, Ramalho F, et al. (2018) Reduced hydroxymethylation characterizes medulloblastoma while TET and IDH genes are differentially expressed within molecular subgroups. J Neurooncol 139: 33-42. [Crossref]
- Varjosalo M, Taipale J (2008) Hedgehog: functions and mechanisms.Genes Dev22: 2454-2472. [Crossref]
- Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW (2016) Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel) 8: E22. [Crossref]
- Epstein EH (2008) Basal cell carcinomas: attack of the hedgehog.Nat Rev Cancer8: 743-754. [Crossref]
- Cheng SY, Bishop JM (2002) Suppressor of fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc Natl Acad Sci U S A 99: 5442-5447. [Crossref]
- Kogerman P, Grimm T, Kogerman L, Krause D, Undén AB, et al. (1999) Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol 1: 312-319. [Crossref]
- Paces-Fessy M, Boucher D, Petit E, Paute-Briand S, Blanchet-Tournier MF (2004) The negative regulator of Gli, suppressor of fused (Sufu), interacts with SAP18, Galectin3 and other nuclear proteins. Biochem J 378: 353-362. [Crossref]
- Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H (1999) Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 126: 3915-3924. [Crossref]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, et al. (2015) Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85: 841-851. [Crossref]
- Ridzewski R, Rettberg D, Dittmann K, Cuvelier N, Fulda S, et al. (2015) Hedgehog Inhibitors in Rhabdomyosarcoma: A Comparison of Four Compounds and Responsiveness of Four Cell Lines.Front Oncol5: 130. [Crossref]
- Xu Y, Xu C, Kato A, Tempel W, Abreu JG, et al. (2012) Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell 151: 1200-1213. [Crossref]
- Kang J, Lienhard M, Pastor WA, Chawla A, Novotny M, et al. (2015) Simultaneous deletion of the methylcytosine oxidases Tet1 and Tet3 increases transcriptome variability in early embryogenesis. Proc Natl Acad Sci U S A 112: E4236-E4245. [Crossref]
- Ramasamy SK, Kusumbe AP, Wang L, Adams RH (2014) Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507: 376-380. [Crossref]
- Wakefield LM, Hill CS (2013). Beyond TGFbeta: roles of other TGFbeta superfamily members in cancer. Nat Rev Cancer 13: 328-341. [Crossref]
- Bierie B, Moses HL (2006) Tumor microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 6: 506-520. [Crossref]
- Watabe T, Miyazono K (2009) Roles of TGF-beta family signaling in stem cell renewal and differentiation.Cell Res19: 103-115. [Crossref]
- Ikushima H, Miyazono K (2010) TGFbeta signalling: a complex web in cancer progression.Nat Rev Cancer10: 415-424. [Crossref]
- Xiao Z, Liu X, Henis YI, Lodish HF (2000) A distinct nuclear localization signal in the N terminus of Smad 3 determines its ligand-induced nuclear translocation. Proc Natl Acad Sci U S A 97: 7853-7858. [Crossref]
- Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, et al. (2015) Targeting the TGFβ pathway for cancer therapy.Pharmacol Ther147: 22-31. [Crossref]
- Gong C, Valduga J, Chateau A, Richard M, Pellegrini-Moïse N, et al. (2018) Stimulation of medulloblastoma stem cells differentiation by a peptidomimetic targeting neuropilin-1. Oncotarget 9: 15312-15325. [Crossref]
- Ye Z, Li J, Han X, Hou H, Chen H, et al. (2016) TET3 inhibits TGF-beta1-induced epithelial-mesenchymal transition by demethylating miR-30d precursor gene in ovarian cancer cells. J Exp Clin Cancer Res 35: 72. [Crossref]
- Gong F, Guo Y, Niu Y, Jin J, Zhang X, et al. (2016) Epigenetic silencing of TET2 and TET3 induces an EMT-like process in melanoma. Oncotarget 8: 315-328. [Crossref]
- D'Souza B, Laurence M, Gerry W (2010) Chapter Three - Canonical and Non-Canonical Notch Ligands. Curr Top Dev Biol 32: 73-129. [Crossref]
- Borggrefe T, Oswald F (2009) The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci 66: 1631-1646. [Crossref]
- Conlon RA, Andrew GR, Janet R (1995) Notch1 is required for the coordinate segmentation of somites. Development 121: 1533-1545. [Crossref]
- Buas MF, Kabak S, Kadesch T (2009) Inhibition of myogenesis by Notch: evidence for multiple pathways.J Cell Physiol218: 84-93. [Crossref]
- Bray SJ (2006) Notch signalling: a simple pathway becomes complex.Nat Rev Mol Cell Biol7: 678-689. [Crossref]
- Pece, S.; et al. (2004) Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol 167: 215-221. [Crossref]
- Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, et al. (2004) Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res 64: 7787-7793. [Crossref]
- Sakaki-Yumoto M, Katsuno Y, Derynck R. (2013) TGF-beta family signaling in stem cells. Biochim Biophys Acta 1830: 2280-2296.? [Crossref]
- Ramasamy SK, Kusumbe AP, Wang L, Adams RH (2014) Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507: 376-380. [Crossref]
- Zhao J, Ma XL, Ma JX, Sun L, Lu B, et. al., TET3 Mediates Alterations in the Epigenetic Marker 5hmC and Akt pathway in Steroid-Associated Osteonecrosis. J Bone Miner Res 32: 319-332. [Crossref]
- Kim M, Park YK, Kang TW, Lee SH, Rhee YH, et al. (2014) Dynamic changes in DNA methylation and hydroxymethylation when hES cells undergo differentiation toward a neuronal lineage. Hum Mol Genet 23: 657-667. [Crossref]
- Terragni J, Zhang G, Sun Z, Pradhan S, Song L, et al. (2014) Notch signaling genes Myogenic DNA hypomethylation and 5-hydroxymethylcytosine. Epigenetics 9: 1-9. [Crossref]
- Li C, Lan Y, Schwartz-Orbach L, Korol E, Tahiliani M, et al. (2015) Overlapping Requirements for Tet2 and Tet3 in Normal Development and Hematopoietic Stem Cell Emergence. Cell Rep 12: 1133-1143.? [Crossref]