Aspirin (Asp) is a member of nonsteroidal anti-inflammation drug and widely used as an analgesic, antipyretic, and anti-inflammation agent. In this investigation, the inherent stability, chemical reactivity, and biological properties of Aspirin and its metabolites have been studied. Density functional theory (DFT) with B3LYP/3-21g has been employed to optimize the structures. Frontier molecular orbital features (HOMO-LUMO gap, hardness, and softness), dipole moment, electrostatic potential and thermodynamic properties (electronic energy, enthalpy, Gibb’s free energy) of these metabolites have been investigated. Molecular docking has been performed against prostaglandin H2 (PGH2) synthase protein 5F19 to search the binding affinity and mode(s) of all compounds. It is found that, all compounds are thermodynamically stable; most of them are chemically more reactive and show better binding affinity than the parent drug. ADMET calculations predict the improved pharmacokinetic properties of all metabolites.
Aspirin, Metabolites, Molecular docking, Pharmacokinetics
Aspirin (Asp) is popularly used as analgesic, anti-pyretic, anti-inflammatory, and anti-platelet agent [1-4]. Recently, Asp and its modified derivatives are using in the treatment of cancer [5-7] stroke [8] , and cardiovascular diseases [9,10]. It inhibit the prostaglandin synthesis by blocking cyclooxygenase [11,12]. It has some common side effects including asthma, kidney and stomach diseases [13]. Previously, some of the major metabolites of Asp are detected and reported by many researchers (Figure 1) [14-16]. Till now, the metabolites formation and their biological action is not completely understood. Attempts have taken to optimize the reported metabolites to understand their biochemical behavior on the basis of quantum mechanical methods. The free energy, enthalpy, dipole moment, HOMO-LUMO gap, and molecular electrostatic potential have been calculated to compare their thermal and chemical behavior. Molecular docking has been performed against human prostaglandin synthase protein 5F19 to predict their binding affinity and modes [17,18]. Pharmacokinetic prediction has been performed to compare their absorption, metabolism and toxicity. The main objective of our investigation was to understand the thermodynamic, molecular orbital, binding affinity, and ADMET properties.
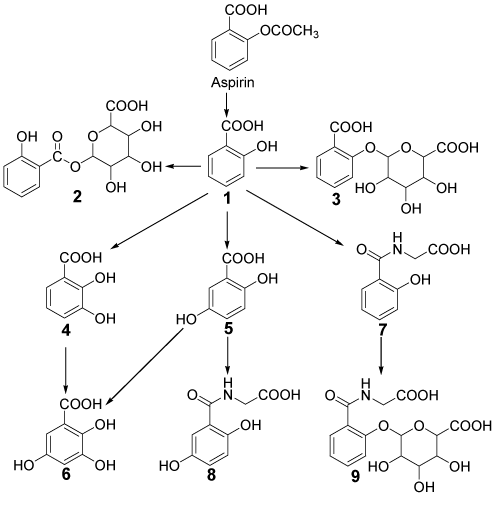
Figure 1. Chemical structures of Aspirin and its major metabolites
1=Salicylic acid, 2= Salicylacyl glucuronide, 3= Salicylphenol glucuronide, 4= 2,3-Dihydroxybenzoic acid, 5= Gentisic acid, 6= 2,3,5-Trihydroxybenzoic acid, 7= Salicyluric acid, 8= Gentisuric acid, 9= Salicyluric phenolic glucuronide
Geometry optimization
In computer aided drug design, quantum mechanical methods are widely used to predict thermal, molecular orbital, and molecular electrostatic potential properties [19]. Initial geometry of Aspirin and its metabolites were taken from the online structure database named ChemSpider [20]. Geometry optimization and further modification of all structures carried out using Gaussian 09 program [21]. Density functional theory (DFT) with Becke’s (B) [22] three-parameter hybrid model, Lee, Yang and Parr’s (LYP) correlation functional [23] under 3-21g basis set has been employed to optimize and elucidate their thermal and molecular orbital properties [24]. Initial optimization of all compounds was performed in the gas phase. Dipole moment, enthalpy, free energy, and electrostatic potential were calculated for all the compounds.
Frontier molecular orbital features HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital) were calculated at the same level of theory. For each of the metabolite, HOMO-LUMO energy gap, hardness (η), and softness (S) were calculated from the energies of frontier HOMO and LUMO as reported considering Parr and Pearson interpretation [25,26] of DFT and Koopmans theorem [27] on the correlation of ionization potential (I) and electron affinities (E) with HOMO and LUMO energy (𝜀). The following equations are used to calculate hardness (η), softness (S);
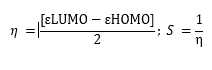
Molecular docking and ADMET prediction
Molecular docking simulation was performed to understand the mechanism of the prostaglandin H2 (PGH2) inhibition of Asp and its metabolites and their binding affinity and mode(s) with target protein [28]. The 3D structure of aspirin acetylated human cyclooxygenase-2 (PDB ID: 5F19) was obtained in pdb format from online protein data bank (PDB) database [29]. All hetero atoms and water molecules were eliminated using PyMol (version 1.3) software packages [30]. Energy minimization of the protein implemented by Swiss-Pdb viewer software (version 4.1.0) [31]. Than optimized drugs were subjected for molecular docking study against human prostaglandin synthase protein (5F19). Finally, molecular docking simulation was performed by PyRx software (version 0.8 ) [32] considering the protein as macromolecule and the drug as ligand. In this analysis, rigid docking was performed where, all rotatable bonds were converted into non-rotatable with the center grid box size 20.8612, 37.5501 and 59.3402 Å along x, y and z directions respectively.
AdmetSAR online database was utilized to predict the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of all metabolites [33].
Thermodynamic analysis
Simple modifications of molecular structure significantly influence the structural properties including thermal and molecular orbital parameters. Spontaneity of a reaction and stability of a product can be predicted from the free energy, and enthalpy values [34]. Highly negative values are more favorable for thermal stability. In drug design, hydrogen bond formation and non-bonded interactions also influenced by dipole moment. Increased dipole moment can improve the binding property [35]. The free energy of Asp is -644.986 Hartree, where the free energies of Salicylacyl glucuronide (2), Salicylphenol glucuronide (3) are almost same (-1174.083 and -1174.103 Hartree respectively), but slightly improved thermal and dipole moment are observed in 3 due to the presence of strong carboxylic (-COOH) group. The highest free energy (-1380.911 Hartree) and dipole moment (8.172 Debye) is observed in Salicyluric phenolic glucuronide (9) due to the presence of a bulky group which suggesting the possible improved binding affinity (Table 1).
Table 1. Molecular formula (MF), molecular weight (MW), enthalpy, free energy in Hartree and dipole moment (Debye) of Aspirin and its metabolites
|
MF |
MW |
Enthalpy |
Free energy |
Dipole moment |
Asp |
C9H8O4 |
180.157 |
-644.936 |
-644.986 |
4.344 |
1 |
C7H6O3 |
138.121 |
-493.187 |
-493.228 |
0.666 |
2 |
C13H14O9 |
314.245 |
-1174.015 |
-1174.083 |
2.856 |
3 |
C13H14O9 |
314.245 |
-1174.035 |
-1174.103 |
5.178 |
4 |
C7H6O4 |
154.120 |
-567.964 |
-568.008 |
4.325 |
5 |
C7H6O4 |
154.120 |
-567.957 |
-568.002 |
3.900 |
6 |
C7H6O5 |
170.120 |
-642.753 |
-642.801 |
5.743 |
7 |
C9H9NO4 |
195.172 |
-699.981 |
-700.033 |
2.435 |
8 |
C9H9NO5 |
211.171 |
-774.784 |
-774.840 |
5.629 |
9 |
C15H17NO10 |
371.296 |
-1380.833 |
-1380.911 |
8.172 |
Molecular orbital analysis
The HOMO-LUMO energies, hardness, softness of all metabolites are presented in Table 2. The electronic absorption relates to the transition from the ground to the first excited state and mainly described by one electron excitation from HOMO to LUMO [36]. The chemical hardness, softness, and chemical potential values depend on the energy of HOMO-LUMO [37,38]. Kinetic stability increase with the increase of HOMO-LUMO gap. As a result, removal of electrons from ground state HOMO to excited state LUMO requires more energy. In this study, the HOMO-LUMO gap of Asp (5.435eV) is greater than its metabolites. The lowest gap (4.263 eV) with highest softness (0.470 eV) is found in Gentisuric acid (Figure 2).
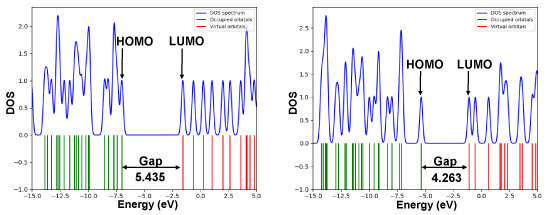
Figure 2. DOS plot and HOMO-LUMO energy gap of Aspirin and Gentisuric acid
Table 2. Energy (eV) of HOMO, LUMO, energy gap, hardness and softness of Asp and its metabolites
|
HOMO |
LUMO |
Gap |
Hardness |
Softness |
Asp |
-6.999 |
-1.564 |
5.435 |
2.717 |
0.368 |
1 |
-6.179 |
-1.352 |
4.827 |
2.414 |
0.414 |
2 |
-6.055 |
-1.370 |
4.685 |
2.344 |
0.427 |
3 |
-6.279 |
-0.965 |
5.314 |
2.657 |
0.376 |
4 |
-5.898 |
-0.979 |
4.919 |
2.460 |
0.407 |
5 |
-5.692 |
-1.122 |
4.570 |
2.285 |
0.437 |
6 |
-5.430 |
-0.981 |
4.449 |
2.225 |
0.450 |
7 |
-6.354 |
-0.937 |
5.417 |
2.708 |
0.370 |
8 |
-5.355 |
-1.092 |
4.263 |
2.132 |
0.470 |
9 |
-6.032 |
-1.095 |
4.937 |
2.468 |
0.405 |
Molecular electrostatic potential
Molecular electrostatic potential (MEP) was calculated to forecast the reactive sites for possible electrophilic and nucleophilic attack of all metabolites [39]. Red colour represent maximum negative area which favourable site for electrophilic attack , blue colour indicate the maximum positive area which favourable site for nucleophilic attack and green colour represent zero potential area. MEP displays molecular size, shape as well as positive, negative and neutral electrostatic potential regions simultaneously in terms of colour grading. From MEP map (Figure 3), region having the negative potential are over electronegative atom (oxygen atoms) and having positive potential are over hydrogen atoms. Here, the potentiality of Asp is -0.211 a.u. and +0.214 a.u. where 9 has the highest potentiality (-0.260 a.u. and +0.215 a.u. respectively), which support the highest possible electrophilic and nucleophilic attack.
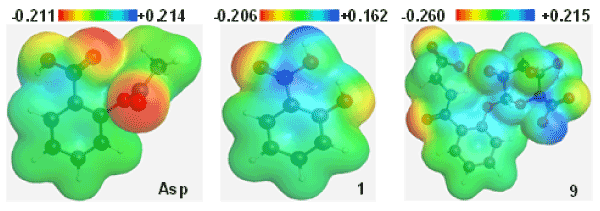
Figure 3. Molecular electrostatic potential map of Asp, 1 and 9
Molecular docking analysis
Binding affinities of metabolites and protein are summarized in Table 3. Greater negative values of binding value indicate stronger binding between drugs and the receptor protein. Strong hydrogen bonding is the most significant contributing factor in increasing binding affinity of drugs with the receptor. Binding affinity and speciality change with the substitution/addition of different functional group. Here, the binding affinity of Asp is 7.0 kcal/mol, improved binding affinities are found for all metabolites except Salicylic acid (1). The binding affinity significantly improved due to the addition of glucuronide (2,3, and 9) and Salicyluric phenolic glucuronide shows the highest binding affinity (-9.5 kcal/mol) (Figure 4).
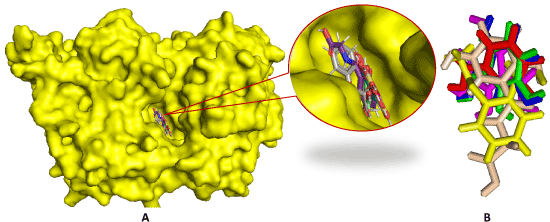
Figure 4. (A) Docked conformation of Asp, 1, 4, 5, 6, and 8 at inhibition bounding site of 5F19
(B). Superimposed view of them after rigid docking
ADMET prediction
ADMET calculation has performed to compare the absorption, metabolism, and toxicity of all metabolites. According to AdmetSAR data (Table 3), Aspirin shows II category acute oral toxicity and rest of the metabolites show III category acute oral toxicity, which suggesting less toxicity of them than the parent drug. All the metabolites are non-carcinogenic, show positive response for blood brain barrier (BBB) and human intestinal absorption criteria. All drugs are P-glycoprotein non-inhibitor where, P-glycoprotein inhibition can interrupt the absorption, permeability and retention of the drugs [40]. However, all the compounds show weak inhibitory feature for human ether-a-go-go-related gene (hERG) which can lead to long QT syndrome [41], so further more study of this aspect is necessary.
Table 3. Binding affinity and selected pharmacokinetic parameters of Asp and its metabolites
Drug |
Binding affinity
(kcal/mol) |
BBB |
Human intestinal absorption |
P-glycoprotein inhibitor |
hERG |
Carcinogen |
Acute oral toxicity |
Asp |
-7.0 |
+(0.945) |
+(0.974) |
NI(0.958) |
WI(0.944) |
NC(0.652) |
II |
1 |
-6.6 |
+(0.616) |
+(0.976) |
NI(0.994) |
WI(0.963) |
NC(0.863) |
III |
2 |
-8.8 |
+(0.691) |
+(0.720) |
NI(0.848) |
WI(0.974) |
NC(0.945) |
III |
3 |
-9.3 |
+(0.691) |
+(0.720) |
NI(0.848) |
WI(0.974) |
NC(0.945) |
III |
4 |
-7.2 |
+(0.623) |
+(0.805) |
NI(0.994) |
WI(0.982) |
NC(0.918) |
III |
5 |
-7.1 |
+(0.666) |
+(0.922) |
NI(0.994) |
WI(0.965) |
NC(0.882) |
III |
6 |
-7.6 |
+(0.622) |
+(0.805) |
NI(0.994) |
WI(0.982) |
NC(0.918) |
III |
7 |
-7.9 |
+(0.576) |
+(0.723) |
NI(0.964) |
WI(0.971) |
NC(0.928) |
III |
8 |
-8.7 |
+(0.576) |
+(0.723) |
NI(0.964) |
WI(0.971) |
NC(0.928) |
III |
9 |
-9.5 |
+(0.781) |
+(0.716) |
NI(0.614) |
WI(0.983) |
NC(0.945) |
III |
NI=Non-inhibitor, WI= Weak –inhibitor, NC= Non-carcinogenic
In this investigation, the inherent stability and biochemical interactions of Asp and its metabolites are studied. All the metabolites have lower HOMO-LUMO gap and have improved pharmacokinetic properties. All the metabolites (except 1) have better binding affinity with the receptor protein and most of them (2,3,7,8, and 9) are thermally more stable than the parent drug. Finally this study may be helpful to understand the thermal, chemical, pharmacological and binding properties of Asp and its metabolites.
We are thankful to Dr. Zainul Abedin Siddique, Professor, Department of Chemistry, University of Chittagong for his valuable suggestions.
Authors declare no conflict of interest.
- Awtry EH, Loscalzo J (2000) Aspirin. Circulation 101: 1206-1218. [Crossref]
- Paez Espinosa EV, Murad JP, Khasawneh FT (2012) Aspirin: pharmacology and clinical applications. Thrombosis 2012: 173124. [Crossref]
- Yeomans ND (2011) Aspirin: old drug, new uses and challenges. J Gastroenterol Hepatol 26: 426-431. [Crossref]
- Fuster V, Sweeny JM (2011) Aspirin: a historical and contemporary therapeutic overview. Circulation 123:768-778
- Thun MJ, Jacobs EJ, Patrono C (2012) The role of aspirin in cancer prevention. Nat Rev Clin Oncol 9: 259-267. [Crossref]
- Patrignani P, Patrono C (2018) Aspirin, platelet inhibition and cancer prevention. Platelets 29: 779-785. [Crossref]
- Patrono C (2019) 50 - Aspirin. In: Michelson ADBT-P (Fourth E (ed). Academic Press, pp 921-936
- Seshasai SRK, Wijesuriya S, Sivakumaran R (2012) Effect of aspirin on vascular and nonvascular outcomes: meta-analysis of randomized controlled trials. Arch Intern Med 172: 209-216
- Capodanno D, Mehran R, Valgimigli M, Baber U, Windecker S, et al. (2018) Aspirin-free strategies in cardiovascular disease and cardioembolic stroke prevention. Nat Rev Cardiol 15: 480-496. [Crossref]
- Ridker PM, Cook NR, Lee I-M (2005) A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med 352:1293-1304
- Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231: 232-235. [Crossref]
- Catella-Lawson F, Reilly MP, Kapoor SC (2001) Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med 345:1809-1817.
- Sostres C, Gargallo CJ, Arroyo MT, Lanas A (2010) Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol 24: 121-132.
- BojiA M, Sedgeman CA, Nagy LD, Guengerich FP (2015) Aromatic hydroxylation of salicylic acid and aspirin by human cytochromes P450. Eur J Pharm Sci 73: 49-56. [Crossref]
- Deng H, Fang Y (2012) Aspirin metabolites are GPR35 agonists. Naunyn Schmiedebergs Arch Pharmacol 385: 729-737
- Li JP, Guo JM, Shang EX, Zhu ZH, Liu Y, et al. (2017) Quantitative determination of five metabolites of aspirin by UHPLC-MS/MS coupled with enzymatic reaction and its application to evaluate the effects of aspirin dosage on the metabolic profile. J Pharm Biomed Anal 138: 109-117. [Crossref]
- Uzzaman M, Uddin MN (2019) Optimization of structures, biochemical properties of ketorolac and its degradation products based on computational studies. DARU J Pharm Sci 1-12
- Uzzaman M, Hoque MJ (2018) Physiochemical, molecular docking, and pharmacokinetic studies of Naproxen and its modified derivatives based on DFT. Int J Sci Res Manag 6: C-2018
- Gleeson MP, Gleeson D (2009) QM/MM calculations in drug discovery: a useful method for studying binding phenomena? J Chem Inf Model 49: 670-677. [Crossref]
- Pence HE, Williams A (2010) ChemSpider: An Online Chemical Information Resource. J Chem Educ 87: 1123-1124.
- Gaussian RA, MJ Frisch, GW Trucks, HB Schlegel, GE Scuseria, et al. (2009) Gaussian. Inc, Wallingford CT.
- Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38: 3098-3100.
- Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37: 785-789.
- Kruse H, Goerigk L, Grimme S (2012) Why the Standard B3LYP/6-31G* Model chemistry should not be used in dft calculations of molecular thermochemistry: Understanding and correcting the problem. J Org Chem 77: 10824-10834.
- Calais JL (1993) Density-functional theory of atoms and molecules. R.G. Parr and W. Yang, Oxford University Press, New York, Oxford, 1989. IX + 333 pp. Price £45.00. Int J Quantum Chem 47: 101.
- Pearson RG (1995) The HSAB Principle — more quantitative aspects. Inorganica Chim Acta 240: 93-98.
- Pearson RG (1986) Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci 83: 8440-8441.
- Seeliger D, de Groot BL (2010) Conformational transitions upon ligand binding: holo-structure prediction from apo conformations. PLoS Comput Biol 6: e1000634. [Crossref]
- Lucido MJ, Orlando BJ, Vecchio AJ, Malkowski MG (2016) Crystal structure of aspirin-acetylated human cyclooxygenase-2: Insight into the formation of products with reversed stereochemistry. Biochemistry 55: 1226-1238.
- Delano WL (2002) The PyMOL molecular graphics system. De-Lano Scientific, San Carlos, CA, USA.
- Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 18: 2714-2723.
- Dallakyan S, Olson AJ (2015) Small-Molecule library screening by docking with PyRx. In: Hempel JE, Williams CH, Hong CC (eds) Chemical Biology: Methods and Protocols. Springer New York, New York, NY, pp 243-250
- Cheng F, Li W, Zhou Y, Shen J, Wu Z, et al. (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52: 3099-3105. [Crossref]
- Cohen N, Benson SW (1993) Estimation of heats of formation of organic compounds by additivity methods. Chem Rev 93: 2419-2438
- Lien EJ, Guo ZR, Li RL, Su CT (1982) Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J Pharm Sci 71: 641-655.
- Saravanan S, Balachandran V (2014) Quantum chemical studies, natural bond orbital analysis and thermodynamic function of 2, 5-dichlorophenylisocyanate. Spectrochim Acta Part A Mol Biomol Spectrosc 120: 351-364.
- Azam F, Alabdullah NH, Ehmedat HM (2017) NSAIDs as potential treatment option for preventing amyloid ß toxicity in Alzheimer’s disease: an investigation by docking, molecular dynamics, and DFT studies. J Biomol Struct Dyn 1-19.
- Parr RG, Zhou Z (1993) Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters. Acc Chem Res 26: 256-258.
- Politzer P, Murray JS (1991) Molecular electrostatic potentials and chemical reactivity. Rev Comput Chem 273-312.
- Amin ML (2013) P-glycoprotein Inhibition for optimal drug delivery. Drug Target Insights 7: 27-34. [Crossref]
- Sanguinetti MC, Tristani-Firouzi M (2006) hERG potassium channels and cardiac arrhythmia. Nature 440: 463-469.