The ETS transcription factors regulate expression of genes involved in normal cell development, proliferation, differentiation, angiogenesis, and apoptosis, consisting of 28 family members in humans. Dysregulation of these transcription factors facilitates cell proliferation in cancers, and several members participate in invasion and metastasis by activating certain gene transcriptions. ETS1 and ETS2 are the founding members of the ETS family and regulate transcription by binding to ETS sequences. Three chimeric genes involving ETS genes have been identified in human cancers, which are EWS-FLI1 in Ewing’s sarcoma, TMPRSS2-ERG in prostate cancer, and ETV6-RUNX1 in acute lymphocytic leukemia. Although these fusion transcripts definitely contribute to the pathogenesis of the disease, the impact of these fusion transcripts on patients’ prognosis is highly controversial. In the present review, the roles of ETS protein translocations in human carcinogenesis are discussed.
ETS, ERG, EWS, FLI1, TMPRSS2, ETV6, RUNX1 expression, cancer
The oncogene v-Ets was discovered as a component of a chimeric gene, along with a truncated v-Myb gene, present in the genome of E26, an avian leukosis virus [1-3]. Subsequently, v-Ets-related genes have been identified from different species. The Ets family has been established as one of the largest group of transcriptional factors, with diverse functions [2, 3]. To date, 28 human ETS family proteins have been identified [3-5]. All ETS genes have a conserved amino acid sequence (the ETS domain) of approximately 85 amino acids that forms the DNA-binding domain at the C-terminus required for the recognition of the consensus core sequence GGAA/T (ETS binding site [EBS]) (Figure 1B). Binding of ETS proteins to target genes is accelerated by the binding of other trans-acting factors to close proximity to the EBS. The pointed (PNT) domain of 65–85 amino acids is the second most conserved region found in a subset of ETS genes (Figure 1B). This domain is found in 11 of 28 human ETS genes and has been shown to participate in protein-protein interaction and oligomerization [3-5]. The human ETS factors are classified into 11 subgroups: ETS (ETS1/2), ERG, FLI1, ETV (PEA3, ETV1/4/5), TEL (ETV6/7), ELG (GABPα), TCF (ELK1/3/4), ELF (ELF1/2/4), SPI1 (SPI1/B/C), ERF (ERF, ETV3, ETV3L), and FEV (Figure 1) [4]. In addition, a subset of four ETS family genes (ELF3, ELF5, EHF, SPDEF) has been placenta-specific subgroup based upon their restricted expression to tissues with high epithelial cell content (4-6); there are total of 28 ETS proteins in humans [4,5].
ETS factors are regulators of the expression of genes that are involved in various biological processes in response to various signaling cascades, cellular proliferation, differentiation, apoptosis, migration, invasion/metastasis [7]. They also control cell adhesion, tissue remodeling, extracellular matrix composition, and angiogenesis [7]. ETS protein functional activity is modulated by post-translational modification and interaction with other nuclear proteins. The importance of ETS genes in human carcinogenesis is supported by the observations that ETS genes have altered expression patterns, amplified, deleted mutated, and most importantly are located at translocation breakpoints in human leukemias and solid tumors [4-6]. Correlation of ETS gene expression levels with tumor progression has been found in most human neoplasias, including leukemias, thyroid, pancreas, liver, prostate, colon, lung, and breast cancers [2]. Aberrant expression of ETS1 and 2 proteins results in the altered regulation of their target response genes, which was recently reviewed [4-6].
The ETS transcriptions factors regulate numerous genes by binding to GGAA/T core of DNA-binding, and are involved in stem cell development, cell proliferation, differentiation, development, transformation, angiogenesis, and apoptosis (Figure 1) [7]. ETS1 and ETS2 are representative members of the ETS family of transcription factors and are downstream effectors of the RAS/RAF/ERK pathway, which involve in cellular proliferation, differentiation, apoptosis and transformation [7].
It was reported that the ETS2 gene was located approximately 17 cM from the breakpoint of common t(8;21) translocation of M2 subtype of acute myelogenous leukemia (AML) [8, 9]. They demonstrated that ERG was situated just proximal to ETS2. However, AML1/ETO was cloned and shown to be responsible for t(8;21) in acute myelogenous leukemias [10,11]. Rao et al. performed in situ hybridization and somatic cell hybridization that the ERG gene is located at chromosome 21q22.3 [12,13]. They found that the ERG gene was translocated from chromosome 21 to chromosome 8 in the t(8;21)(q22;q22). It was then found that ERG2 is a nuclear phosphoprotein that bound to purine-rich sequences [14]. ERG is considerably more stable than the short-lived ETS1 and ETS2 proteins with a half-life of 21 hours [7]. ERG2 is a sequence-specific, DNA-binding protein and is expressed at higher levels in early myeloid cells than in mature lymphoid cells, acting as a regulator of genes required for maintenance and differentiation of early hematopoietic cells.
The ETS family gene often fuses with other genes as a result of translocation. The ERG gene is found at the breakpoint of t(16;21)(p11;q22) in the FUS:ERG fusion in leukemia [15]. In solid tumors, the ERG gene is activated by translocation in Ewing sarcoma with EWS:ERG translocation t(11;22)(q24,q22) [16] and EWS:FLI1 translocation t(21,22)(q22,q12) (Fli1 belongs to ERG family of proteins) (Figure 1B) [4].
It has been reported that the TMPRSS-ERG translocation is found in 50% of prostate cancer (PCa), and is associated with prognosis (Figure 1B) [17-19]. Studies suggest the involvement of another member of the ETS family, known as ETV6, in human cancer. ETV6 is frequently activated in human leukemia with ETV6-RUNX1 (TEL-AML1) [20,21]. The roles of these ETS proteins in causing human cancer, especially acute leukemia, Ewing’s sarcoma, and PCa are discussed in this review.
Gene cloning and expression
The Reddy’s group isolated a cDNA that comes with the complete coding sequence of ERG and predicted 363-residue protein. They predicted that ERG share about 40% and 70% homology with the 5’ and 3’ regions of the v-Ets oncogene, respectively, suggesting that ERG belongs to the ETS oncogene family (Figure 1B) [22]. Through sequencing analysis, the same group identified 2 different ERG transcripts, ERG1 and ERG2. ERG2 differs from ERG1 by a splicing event that causes a frameshift, resulting in an additional 99 amino acids at the N terminus [22]. They found that alternative sites of splicing and polyadenylation, together with alternative sites of translation initiation, allow synthesis of 2 ERG polypeptides, ERG1 and ERG2. Then Owczarek et al. reported that 5 alternatively spliced ERG transcripts encode proteins of 38 to 55 kD, all of which bind to ETS sites of genomic DNA and acted as transcriptional factors [23]. RT-PCR analysis of 6 ERG variants showed variable expression in placenta and most human cell lines examined. They found that the ERG gene contained at least 17 exons that have extension of >280 kb.
EWS:ERG fusion gene in Ewing sarcoma
The ERG gene often fuses with others as a result of translocation as described earlier. Ichikawa et al. demonstrated that in human myeloid leukemia with t(16;21)(p11;q22), the TLS/FUS gene on chromosome 16 was fused with ERG gene on chromosome 21 [24]. The chimeric FUS:ERG gene product is an DNA-binding protein that was highly homologous to the product of the EWS gene involved in Ewing sarcoma. FUS and EWS are homologous to each other. Thus, the FUS:ERG gene fusion found in t(16;21) leukemia was predicted to produce a protein similar to the EWS:ERG chimeric protein seen in Ewing's sarcoma.
Ewing sarcoma, the second most common malignant bone tumor of children and young adults, is an aggressive osteolytic tumor with a marked propensity for dissemination to the bones (arms, ribs, legs, pelvis), lungs, spine, and bone marrow [25-27]. It belongs to the heterogeneous group of small round cell tumors that are often a diagnostic challenge to the pathologist and clinical oncologist. EWS and related peripheral primitive neuro-ectodermal tumors show a t(11;22)(q24,q22) translocation (present in 85% of Ewing's sarcoma; EWS-FLI1 fusion) [28] or a t(21,22)(q22,q12) (EWS-ERG fusion) that is associated with hybrid transcripts of the EWS gene with the FLI1 or ERG gene [29]. The chimeric protein resulting from the fusion of EWS with ERG is structurally similar to the typical EWS-FLI1 protein in which the N-terminal portion of EWS is linked to the ETS domain of ERG (Figures 1 and 2) [29-33]; Upon fusion, EWS-FLI1 becomes a more potent transactivator than FLI1 itself although it does not transactivate the Arf promoter [35]. It has been reported that many targets of EWS-FLI1, both direct and indirect, have been found to be involved in Ewing sarcoma maintenance [33]. EWS-FLI1 collaborates with several proteins to modulate mRNA transcription and splicing (Figure 2) [29-33]. Biochemical and computer studies have revealed that the EWS-FLI1 molecule is intrinsically disordered, from the amino-terminus of EWS to the DNA-binding domain of FLI1, and that unique side chains within a structure of EWS-FLI1 are critical for the transcription and transforming activity of the EWS family proteins (Figure 2) [34]. Interestingly it was reported that EWS-FLI1 employs an E2F switch to drive target gene expression [35-39]. The Bayesian model (a theory in the field of statistics in which the evidence about the true state of the world is expressed in terms of degrees of belief) revealed that the formation of a synergistic complex between EWS-FLI1 and E2F3 was the most likely mechanism explaining the observed kinetics of E2F target induction [36]. They proposed that aberrant cell cycle activation in EWS was due to the physical recruitment of E2F3 by EWS-FLI1 replacing E2F4 on their target promoters [36].
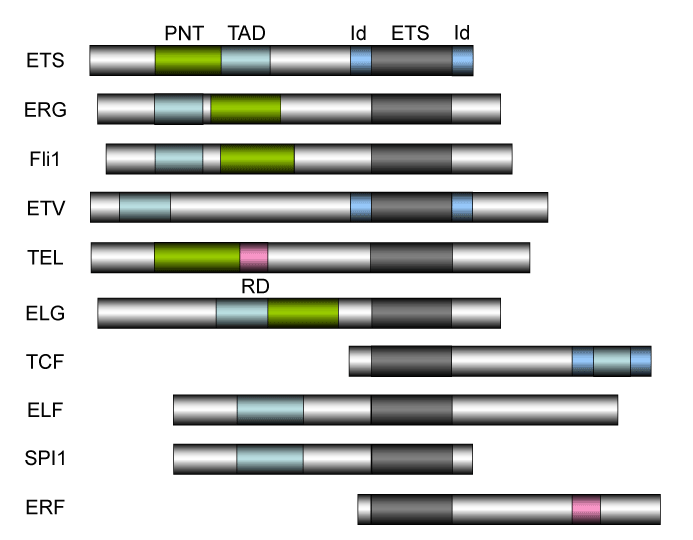
Figure 1. The structure of ETS family proteins
The structure of ETS family proteins. The human ETS factors are classified into 11 subgroups: ETS (ETS1/2), ERG, FLI1, ETV (PEA3, ETV1/4/5), TEL (ETV6/7), ELG (GABPα), TCF (ELK1/3/4), ELF (ELF1/2/4), SPI1 (SPI1/B/C), ERF (ERF, ETV3, ETV3L), and FEV [4,5,165,166]. There are totally 28 ETS proteins in humans. ETS translocations involving Fli1, ERG, and TEL have been discussed in this review. The ETS domain that is essential for DNA-binding is shown in dark box. The DNA-binding by the ternary complex factor (TCF) subfamily of ETS-domain transcription factors is tightly regulated by intramolecular and intermolecular interactions [167-169]. The helix-loop-helix (HLH) - containing Id proteins are trans-acting negative regulators of DNA binding by the TCFs. Id domains have been identified in ETS, ETV, and TCF. PNT: pointed domain, TAD: transactivation domain, ID: Id-interaction domain, ETS: ETS domain (DNA-binding domain), RD: ring domain.
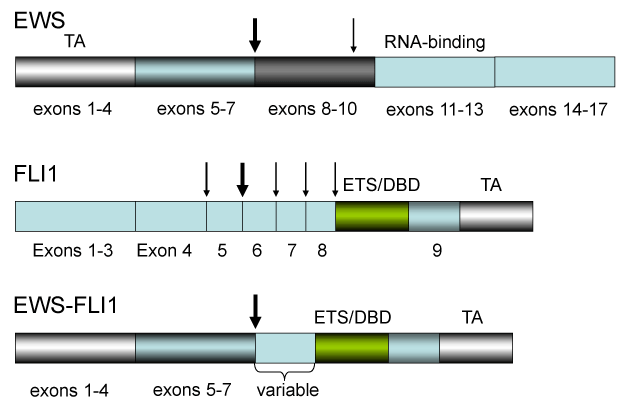
Figure 2. The EWS-FlI1 chimeric gene found in Ewing’s sarcomas
The EWS-FlI1 chimeric gene is found in 80% of Ewing sarcomas. Vertical arrow shows the major breakpoints (between exon 7 and 8 in EWS gene and exons 5 and 6 in ETS-like FLI1 gene). The thin arrows represent the minor breakpoints. The EWS-FLI1 gene is a fusion between EWS and FLI1, transactivation (EWS) and DNA-binding (FLI1) domains. The structure between EWS and FLI1 varies dependent on the Ewing’s sarcoma.
A direct transcriptional target of EWS-FLI1 is NROB1/DAX1 and it is a key effector in EWS-FLI1-mediated oncogenesis [40]. siRNA-mediated depletion of DAX1 in EWS-FLI1 cell lines results in growth arrest and inhibition of tumor formation in immunodeficient mice. DAX1 also physically interacts with EWS-FLI1 to control gene expression and to modulate the transformed phenotype of Ewing sarcoma [40,41]. Other known direct transcriptional targets of EWS-FLI1 are glutathione-S-transferase M4, protein tyrosine phosphatase L1 (PTPL1), GLI1, and AP1 [42-44]. Similar to the inhibition of PTPL1, pharmacological inhibition of GLI1 decreased proliferation and soft agar colonies in Ewing sarcoma family tumor (ESFT) cells. In many cases, however, pathogenesis of EWS-FLI1 depends on cooperative DNA-binding with other transcription factors, such as AP1, which have tandem binding sites close to the EWS-FLI1 target promoters [45].
Pathogenesis of Ewing’s sarcoma also depends on other genetic alterations or cellular functions that indirectly control EWS-FLI1 activity. Among these functions are the p53 and RB pathways, hypoxia, IGF1/IGF1R signaling, and microRNAs [46]. Loss of tumor suppressor genes, such as p53 [47,48] or Ink4a/ARF [49,50], greatly accelerate tumorigenesis in EWS-FLI1-transgenic mice [51,52], and studies have shown that loss of p16Ink4a stabilizes EWS-FLI1 expression and cooperates with EWS-FLI1-mediated transformation [53]. Although EWS-FLI1 recognizes the ETS sequence on the Arf promoter, it does not affect the Arf transcription like the Myb-like protein DMP1 [35,37,54-73]. These data indicate that p16Ink4a is the target in the pathogenesis of EWS. In ESFT, hypoxia has been shown to contribute to resistance of apoptosis via HIF1α [74]. EWS-FLI1 expression increases transiently under hypoxic conditions in a HIF1α-dependent manner; colocalization of HIF1α and necrotic areas in an ESFT tissue array has been reported [74].
Transgenic mouse models have shown their ability to closely reproduce the conditions of transgenes in mice [75,76]. However, no genetically engineered mouse models (GEMM) have been created for Ewing’s sarcoma so far because transgenic expression of the EWS-Fl1 fusion gene causes embryonic lethality [52]. The differences in signaling pathways, Ets-binding sites, co-regulators between mice and humans mice cause additional issues. To make GEMM for EWS, EWS-FLI1 should be expressed exclusively in mesenchymal stem cells (MSCs) to avoid its expression in hematological tissues [77]. Torchia et al. therefore took an approach to express EWS-FLI1 in mesenchymal tissues. They created mice with Cre-inducible expression of EWS-FLI1 from the ubiquitous Rosa26 locus [78]. When crossed with Mx1-Cre mice (interferon-inducible the most commonly used “deleter strain” in experimental hematology that is interferon-inducible) [79], activation of EWS-FLI1 resulted in the rapid development of myeloid/erythroid leukemia characterized by expansion of primitive mononuclear cells causing hepatosplenomegaly, severe anemia, and death [78]. Gene expression profiles of primary and transplanted animals were highly similar, suggesting that activation of EWS-FLI1 was the primary event leading to disease in this model. The Cre-inducible EWS-FLI1 mice provided a novel model system to study the contribution of this oncogene to malignant disease in vivo [78].
The Lozano lab created conditional transgenic GEMM for Ewing’s sarcoma by inducing EWS-FLI1 expression in the primitive MSC of the embryonic limb buds by making use of specific recombinases that allow spatio-temporal control of the EWS-FLI1 gene (EF mice) [52]. When crossed to the Prx1-Cre transgenic mouse, which expresses Cre recombinase in the primitive mesenchymal cells of the embryonic limb bud, the EF mice had number of developmental defects of the limbs [52]. These included shortening of the limbs, muscle atrophy, cartilage dysplasia, and immature bone. Under the condition of p53 deletion, EWS-FLI1 accelerated the formation of sarcomas from a median time of 50 to 21 weeks, with poorly differentiated phenotype [52]. Taken together, the results suggest that EWS-FLI1 inhibits normal limb development and accelerates the formation of poorly differentiated sarcomas.
One important issue we have to remember to reproduce Ewing’s sarcoma in GEMM is the influence of the reciprocal chimeric protein FLI1-EWS generated from the locus [80]. Elzi et al. reported that FLI1-EWS was frequently expressed in Ewing’s sarcoma and presented evidence that endogenous FLI1-EWS was required for Ewing’s sarcoma growth, and that it cooperated with EWS-FLI1 in human mesenchymal stem cells through abrogation of the proliferation arrest induced by EWS-FLI1. Thus, creation of double transgenic GEMM for EWS-FLI1 and FLI1-EWS under specific promoter might be necessary to reproduce Ewing’s sarcoma [80].
TMPRSS2-ERG fusion gene in prostate cancer (PCa)
By using a bioinformatics approach (Figure 3), Tomlins et al. [81,82] reported candidate oncogenic chromosomal aberrations in PCa. Two ETS transcription factors, ERG and ETV1, were identified (Figure 1). They demonstrated that 23 of 29 PCa samples (79%) had rearrangements in ERG or ETV1 by FISH. Later experiments have shown that the TMPRSS2-ERG translocation was found in ~50% of Caucasians, with a lower frequency in African Americans, and even less common in Asians [83]. Cell line experiments suggested that the androgen-responsive promoter elements of TMPRSS2 mediate the overexpression of ETS family members in PCa [81]. Using chromatin immunoprecipitation coupled with sequencing, Yu et al. found that ERG disrupts androgen receptor (AR) signaling by inhibiting AR expression, binding to and inhibiting AR activity, and inducing repressive epigenetic programs via direct activation of the H3K27 methyltransferase EZH2 [84]. In normal tissues, the TMPRSS2 and ERG genes are localized tandemly on chromosome 21q22 (Figure 3) [81]. The TMPRSS2-ERG fusion joins TMPRSS2 exons 1 or 2 to ERG exons 2, 3, or 4, which results in activation of the ERG transcription factor [81,85].
The mechanism of translocation was studied by Mani et al. [86]. By studying human PCa cells with fluorescence in situ hybridization, they showed that androgen signaling induces proximity of the TMPRSS2 and ERG genomic loci, both located on chromosome 21q22.2 (Figure 3). Subsequent exposure of the cells to γ-irradiation, which causes DNA double-strand breaks, facilitates the formation of the TMPRSS2-ERG gene fusion. Their results explain why TMPRSS2-ERG gene fusions are restricted to the prostate, which is dependent on androgen signaling [86].
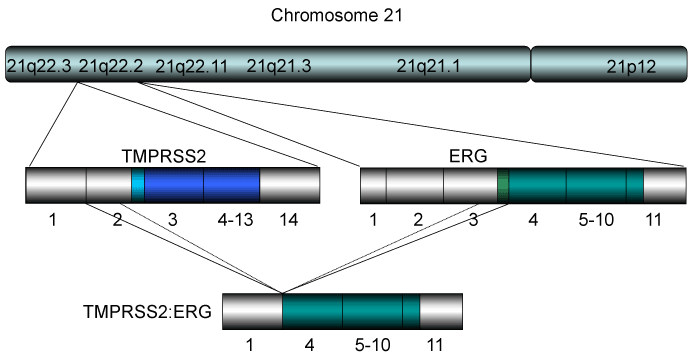
Figure 3. The TMPRSS2-ERG gene found in prostate cancer
The TMPRSS2-ERG or ETV chimeric gene has been found in ~80% of prostate cancer samples of radical prostatectomy [81]. The former translocation has been found in ~50% of Caucasians, with a lower frequency in African Americans, and even less common in Asians [83]. The structure of chromosome of 21 where the chimeric gene for TMPRSS2-ERG is derived is shown. Most of the resultant TMPRSS2-ERG gene consists of exon 1 of TMPRSS2 (non-coding) and exons 4-11 of ERG indicating that the gene makes near full-length proteins of ERG consistent with ERG overexpression in PCa. On the other hand, the TMPRSS2-ETV1 fusion results in chimeric protein that consists with a part of TMPRSS2 and truncated form of ETV1, resulting in expression of the fusion protein that is not expressed in normal tissues.
Transgenic models for TMPRSS2-ERG gene fusion in mice
Tomlins et al. [87] studied the role of TMPRSS2-ERG gene fusion product using in vitro and in vivo model systems. Transgenic mice expressing the ERG gene fusion product under androgen-regulation developed PIN. Introduction of the ERG gene fusion product into primary or immortalized benign prostate epithelial cells induced an invasion-associated transcriptional program, but did not increase cellular proliferation or anchorage-independent growth [87]. ERG knockdown in VCaP cells (PCa cell line established from a vertebral metastatic lesion) induced a transcriptional program consistent with prostate differentiation [87]. Importantly, VCaP cells and benign prostate cells overexpressing ERG directly engage components of the plasminogen activation pathway to mediate cellular invasion, potentially representing a downstream ETS target susceptible to therapeutic intervention. Thus, TMPRSS2-ERG fusions mediate invasion, consistent with the histologic distinction between PIN and PCa [87].
Carver et al. [88] reported that aberrant expression of ERG is a progression event in prostate tumorigenesis. Interestingly, PCa specimens containing the TMPRSS2-ERG rearrangement were significantly enriched for loss of the tumor suppressor PTEN, consistent with the report by King et al. [89]. Similarly, transgenic overexpression of ERG in mouse prostate tissue promotes marked acceleration and progression of high-grade prostatic PIN to prostatic adenocarcinoma in a Pten-heterozygous background [88]. In vitro overexpression of ERG promotes cell migration, a property necessary for tumorigenesis, without affecting proliferation. They showed that ADAMTS1 and CXCR4, two candidate genes strongly associated with cell migration, were upregulated in the presence of ERG overexpression [88]. Thus, ERG has a distinct role in PCa progression and cooperates with PTEN haploinsufficiency to promote progression of high grade PIN to invasive adenocarcinoma.
Recently, Mounir et al. compared the transcriptional effects of TMPRSS2-ERG expression in a transgenic mouse model [90]. ERG repressed the expression of a previously unreported set of androgen receptor (AR)-independent neuronal genes that were indicative of neuroendocrine (NE) cell differentiation. Cell sorting and proliferation assays performed after ERG knockdown indicated that ERG drove proliferation and blocked the differentiation of prostate cells to both NE and luminal cell types [90]. They also provided evidence that these NE cells were resistant to pharmacological AR inhibition, and can revert to the phenotype of parental cells upon restoration of AR/ERG signaling. Thus ERG may have a direct role in preventing resistance to anti-androgen therapy [90].
Expression of the TMPRSS2-ERG fusion protein in PCa
Although the TMPRSS2 (chr 21q22.3, exon 1) - ERG (chr 21q22.2, exon 4) fusion with intra-chromosomal deletion of 3MB on chromosome 21 is quite common (Figure 3), other expressed variants have been reported [91,92] differing in the exon break points in TMPRSS2 (exons 1-5) and ERG (exons 2-5). Depending on the exon break point, protein translation is predicted to start either (1) at the genuine ATG of ERG (exon 3), (2) the native ATG of TMPRSS2 (exon 2), or (3) an in-frame ATG within exon 4 or 5 of ERG; in the third cases the N terminus of ERG would be truncated (Figure 3). This is important since the expression of selected TMPRSS2-ERG break point variants has recently been linked to specific clinicopathological features of PCas.
Wang et al. [93] reported that the expression of TMPRSS2-ERG fusion mRNAs and correlated the isoforms expressed and expression levels with clinical outcome in cancers from men undergoing radical prostatectomy. Overall, 59% of clinically localized PCas expressed the TMPRSS2-ERG fusion gene, confirming the initial observations of high frequency expression of this mRNA in PCa [81]. Expression of an isoform, in which the native ATG in exon 2 of the TMPRSS2 gene was in frame fused with exon 4 of the ERG gene, was associated with clinical and pathologic manifestations of aggressive disease [93]. Expression of other isoforms, in which the native ERG ATG in exon 3 was used, was associated with seminal vesicle invasion with poor outcome following radical prostatectomy [93]. Cancers not expressing these isoforms expressed higher levels of fusion mRNAs, which was present in PCa with early prostate-specific antigen recurrence. Thus, both isoforms of TMPRSS2-ERG fusions expressed and expression levels might affect PCa progression with poor prognosis [93]. Further studies are needed to elucidate the roles of the known and variant ERG fusions in the pathogenesis and androgen dependency of PCa.
Aberrant TMPRSS2-ERG signaling in PCa
It has been reported that increased expression of ETS proteins in adult murine prostate epithelial cells is sufficient to induce epithelial hyperplasia and focal prostatic PIN lesions, but not enough in progression to carcinoma [94,95], which are consistent with the findings by Tomlins et al. for the fusion protein PCa [81,82]. ERG interacts with activations in PI3K signaling, such as PTEN inhibition or AKT1 upregulation, to cause the development of a well-differentiated adenocarcinoma. Thus, loss of PTEN and the presence of the TMPRSS2-ERG gene fusion are events significantly associated with PCa [96]. Bismar et al. proposed the hypothesis that PCa development might be driven initially by PTEN hemizygous loss that caused high grade PIN lesions, which leads to genomic instability, chromosomal rearrangement, and progression to cancer [97]. Subsequent biallelic PTEN inactivation characterizes a particularly aggressive subset of metastatic and hormone-refractory PCa [98]. HDAC1 upregulation is common in PCa, and was found to be increased in tumors with ERG rearrangement [99]. ERG overexpression in PCa is highly implicated in promoting motility and invasiveness, high levels of HDAC1, and subsequent down-regulation of HDAC1-targeted genes, activation of WNT/β-catenin signaling pathway, and inhibition of apoptosis. Activation of the AR through the WNT/β-catenin signaling results in increase in AR expression, enhanced transcription of TMPRSS2-ERG, and high levels of ERG [100]. Elevated ERG, in turn, modulates the growth of PCa cells by upregulating the MYC oncogene, and by abrogating the differentiation of prostate epithelium [101]. Elevated MYC expression in primary PCa is biologically relevant and may be a predictor of future biochemical recurrence [102]. In summary, activation of the HDAC1-WNT/β-catenin-MYC pathway by ERG arrangement is associated with invasive behavior in PCa [103,104].
TMPRSS2-ERG gene fusion as a prognostic marker for PCa
The prognostic implications of TMPRSS2-ERG fusion gene in PCa have not been established. Some authors found a correlation between the presence of fusion gene/protein and poor prognosis [92,93,105-110], but others found a correlation with good prognosis [111-113], the third group reported no correlation with prognosis of PCa [114-117].
In the first group, Attard et al. reported that patients with two or more copies of the TMPRSS2-ERG fusion gene due to the interstitial deletion had worse survival rates than patients without TMPRSS2-ERG rearrangement [106]. This is consistent with the view that ERG overexpression is responsible for driving cancer progression [118], and that the 2.8 Mb deletion (containing genes with tumor suppressor activity) may contribute to the oncogenic potential of the TMPRSS2-ERG fusion product [81]. To understand molecular signatures associated with poor outcome in PCa, Markert et al. [107] analyzed a microarray data set characterizing 281 PCa from a Swedish cohort. They identified a subset of tumors manifesting stem-like signatures together with p53 and PTEN inactivation, which had very poor survival outcome; a second group characterized by the TMPRSS2-ERG fusion with poor survival outcome, composed of 18% of the tumors in the dataset [107]. Font-Tello et al. reported that the expression levels of the TMPRSS2-ERG fusion and ERG mRNA were associated with high Gleason scores, aggressive PCa phenotypes, low PTEN expression, and poor prognosis [108,109]. Finally Deplus et al. reported that TMPRSS2-ERG increased bone tropism of PCa cells and metastasis development [110]. Overall, these results suggest that TMPRSS2-ERG could be molecular markers of aggressiveness for PCa.
In the second group, Saramäki et al. [111] studied the presence of the TMPRSS2-ERG rearrangement by reverse transcription-PCR and fluorescence in situ hybridization in 19 PCa xenografts and 7 PCa cell lines. The expression of ERG was studied in the xenografts, cell lines, and in 49 freshly frozen clinical prostate samples by quantitative reverse transcription-PCR. Seven of 19 (37%) of the xenografts overexpressed ERG and had TMPRSS2-ERG rearrangement. In clinical tumor specimens, the overexpression of ERG was associated with the rearrangement. Fifty of 150 (33%) of the prostatectomy specimens and 28 of 76 (37%) of the hormone-refractory PCa on tissue microarrays carried the TMPRSS2-ERG rearrangement [111]. It was associated with longer progression-free survival in patients treated by prostatectomy, and was an independent predictor of favorable outcome. The fusion was not associated with other clinical markers such as Gleason score, clinical stage, prostate-specific antigen.
In the third group, Gopalan et al. [115] analyzed TMPRSS2-ERG gene rearrangement status by fluorescence in situ hybridization in 521 cases of clinically localized surgically treated PCa with 95 months of median follow-up. 42% of primary tumors and 40% of metastases had rearrangements. 11% had copy number increase of the TMPRRS2-ERG region. Rearrangement alone was associated with lower grade, but not with stage, biochemical recurrence, metastases, or survival. They reported that a subgroup of cancers with chromosomal number instability (CNI) and rearrangement by deletion, with two or more copies of the deleted locus, tended to be more clinically aggressive. DNA index assessment revealed that the majority of tumors with CNI of TMPRSS2-ERG had generalized aneuploidy/tetraploidy in comparison to control tumors. They concluded that translocation of TMPRSS2-ERG was not associated with clinical outcome [115]. It has also been reported that TMPRSS2-ERG gene fusion was not associated with PCa recurrence, but that ERG gene copy number gain without fusion was associated with twice the risk of PCa recurrence [116, 117]. They explained that the ERG gene copy number increase in PCa was the result of tumor aneuploidy, which potentially leads to chromosome 21 polysomy [118]. In this context, the association of greater probability of tumor progression with increased copy number of the ERG gene formed without fusion is not surprising given the previous evidence supporting aneuploidy as a negative prognostic indicator in PCa [119, 120].
It seems that early studies tended to interpret TMPRSS2-ERG gene fusion as a marker of poor prognosis; however, later large cohort studies report that it is a marker of better prognosis. There are several explanations for the different prognostic values of TMPRSS2-ERG: they are clinical settings, size of patient cohorts, differences in sample collection, and the technique used for determination of fusion gene. It is generally believed that high expression of TMPRSS2-ERG fusion transcript in PCa tumor tissue in combination of high PSA and low PTEN levels are associated with aggressiveness of the tumor with poor prognosis.
Acute lymphoblastic leukemia (ALL) is a hematopoietic malignancy characterized by clonal proliferation of immature blood cell precursors (blasts) that progresses rapidly into systemic disease. Signs of leukemia include fever and hemorrhage caused by reduced number of platelets. In pediatric B-cell ALL, the most frequent chromosomal lesion is t(12;21)(p13;q22), which results in the fusion of ETV6/RUNX1 (E/R fusion; also known as TEL/AML1) fusion gene (Figure 4) [121-123]. This genetic alteration occurs in ~25% of pediatric ALL diagnosed between the ages of 2 and 10 years [123,124]. It was originally believed that this rearrangement is a favorable prognostic indicator based on the excellent molecular response to treatment and beneficial clinical outcome [125-127]. However, others found late relapses occurring in up to 20% of patients [128,129], suggesting that it is a sign of ominous prognosis in some patients.
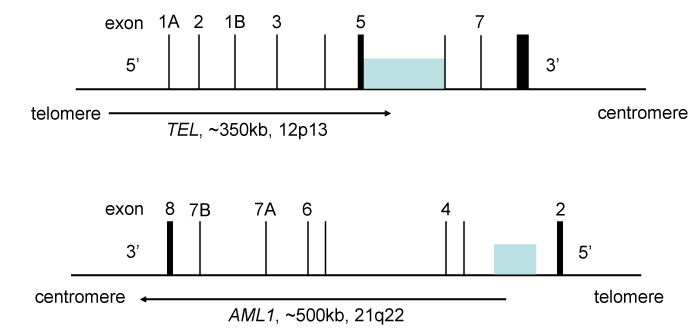
Figure 4. The genomic DNA structure for the TEL-AML1 (ETV6-RUNX1) locus
Translocations involving these loci are found in 25% of pediatric ALL, and are associated with relatively favorable prognosis. The TEL (ETV6) gene is chromosome 12p13, and the AML1 (RUNX1) gene is located on chromosome 21q22. The shaded boxes indicate the locations of breakpoints found in ALL. The numbers are the positions of exons. The breakpoints come from references 168 and 169. The intron 2 in reference 168 is intron 1 in reference 169.
The E/R fusion gene develops as an early event in childhood ALL. The expression of E/R results in generation of a persistent pre-leukemic clone, which postnatally changes to ALL after secondary genetic events [129]. The detection of the E/R fusion sequence in identical twins and pediatric ALL indicates that this gene fusion happens in the prenatal period [130]. However, 1) the occurrence of ALL simultaneously is found in only 5% of twin children, 2) the postnatal latency period of the disease is highly variable, and 3) data from transgenic animal models of E/R fusion [131] suggest that additional genetic events are required for the development of overt ALL. Early studies addressing secondary genetic changes have focused on the deletion of the non-rearranged ETV6 allele which is a tumor suppressor by dimerizing with E/R to reduce its transforming activity [132,133]. Consistently, loss of ETV6 is found in as many as 70% of t(12;21) (+) ALLs [134,135]. Moreover, ~20% of t(12;21) (+) patients have additional genetic change in ETV6 or RUNX1 [136]. These genomic changes are so common that ETV6 has been attributed to the development of E/R(+) ALLs.
To understand the genetic evolution of E/R(+) ALL without prejudice, identification of entire spectrum of genetic changes that accompany this fusion gene is necessary. The genome of E/R (+) ALL has been well-characterized at the copy number and cytogenetic level. Generally, E/R (+) ALL has an average of 2.8 (0-14) additional copy number alterations (CNAs) [137]. As predicted, deletion of 12p (39%) was the most common abnormality resulting in the loss of wild type ETV6 allele [138]. The 9p deletion containing the INK4b/ARF/INK4a locus [139-141] can be seen in up to 25% of E/R(+) patients, and the B-cell differentiation regulator PAX5 [126,142]. CDKN2a/b, ETV6, PAX5, deletions, chromosome 6q loss, and chromosome 16 gain are probably among the earliest genetic aberrations in E/R(+) ALL [143]. Fuka et al. addressed the issue by conducting a shRNA-mediated knock-down (KD) of the E/R fusion gene and investigated the consequences with two E/R(+) leukemic cell lines [144]. Microarray analyses identified 777 genes whose expression was significantly altered. The E/R KD - upregulated set was enriched for genes included in the "cell activation", "apoptosis", "signal transduction", "immune response", and "development and differentiation" categories, whereas in the E/R KD - downregulated set consisted only with the "PI3K/AKT/mTOR signaling" and "hematopoietic stem cells", which was confirmed with primary E/R(+) ALL samples [144]. The results suggest that the E/R chimeric protein accelerates tumorigenesis by up-regulating genes involved in cell proliferation without sending signals to the PI3K-AKT-mTOR pathway.
Pathogenesis, diagnosis, and treatment of ALL in relapse
Most ALL relapses occur during treatment, following discontinuation of treatment or within the first 2-year after treatment completion. Certain relapses have been reported to occur even after longer period of time following the initial diagnosis [125-129,145]. Site of relapse and length of first remission are the major criteria for the classification of patients after first relapse, which include patients with isolated or concurrent marrow, isolated central nervous system (CNS), isolated testicular and other extramedullary relapses with or without CNS involvement [145]. Leukemia relapse is the result of outgrowth of a clonal cell population not entirely eradicated by treatment. Each case of ALL comprises of a unique rearrangement of immunoglobulin or T-cell receptor (TCR) genes; therefore, detailed study on rearrangements of immunoglobulin or TCR genes typical of ALL clones may help to determine the origin of leukemia recurrence. Origin of the relapsed ALL has been addressed based on the study of genome-wide DNA copy number analysis on matched samples obtained at the time of diagnosis and relapse [146].
Standard recovery regimens for relapsed ALL are typically based on different combinations of the same agents used in frontline chemotherapy [146,147]. Many practitioners adopt therapeutic strategies involving risk-adapted treatment, alternating short term chemotherapy, in some cases together with cranial/craniospinal irradiation, and conventional maintenance therapy [146]. Salvage treatment after ALL relapse involves inducing a second remission with conventional intensive chemotherapy and consolidation, re-intensification and maintenance therapy, or allogeneic stem-cell transplantation (SCT) to strengthen a treatment [148]. Effective treatment of ALL relapse very much depends on the risk-based treatment allocation of patients in order to maximize response to therapy while minimizing toxicity and adverse effects from chemotherapy [146-148]. Using the prognostic factors such as first remission duration; site and immunophenotype of relapse; genetic alterations; and initial response to relapse therapy, distinct subgroups of relapsed ALL can be identified, which may either be treated with chemo and radiation therapies or by additional allogeneic steam cell treatment [149]. Since responses to single agent therapy have been poor, integrating new chemotherapy agents in combination with novel approaches, new antimetabolites, and monoclonal antibodies against leukemia-associated antigens can been implemented [150,151].
Molecular mechanisms and the treatment of E/R(+) ALL in relapse
In most E/R patients, relapses occur several years after discontinuation of treatment meaning that relapse is a genetic alterations [125-129]. To characterize the clonal origins of E/R(+) ALL relapse, numerous studies have compared immunoglobulin/T cell receptor gene rearrangements, genomic structure of un-rearranged ETV6 allele, and CNA patterns of diagnostic and relapse samples in the same patient [152-155]. Most importantly, late relapsing cases of E/R(+) ALL are chemosensitive with long-term remissions, implying that initial survival and re-emergence have common properties [156]. Comparison of genomic boundaries of un-rearranged ETV6 allele indicated that the relapse clone derived from a sibling clone already present at diagnosis [153,154]. It is possible that this minor population shows only moderate reduction during initial therapy, but rapidly expands before relapse. After relapse, these clones are rapidly eradicated by chemotherapy [152]. In conclusion, the relapse clones of E/R(+) leukemia originate from those exist at initial presentation.
Then what is the impact of E/R fusion in patients’ responsiveness to therapy? To gain insight into the relapse mechanisms, Kuster et al. [157] analyzed single nucleotide polymorphism arrays for CNAs in 18 matched diagnosis and relapse ALLs. They identified recurrent, mainly non-overlapping deletions associated with glucocorticoid-mediated apoptosis targeting the Bcl2 modifying factor (BMF), glucocorticoid receptor NR3C1, and components of the mismatch repair pathways [157]. Fluorescence in situ hybridization demonstrated that BMF deletions, NR3C1 and mismatch repair alterations became more common at relapse. These findings implicate that glucocorticoid-associated drug resistance in E/R(+) relapse, which is a direction for future therapies.
Gandemer et al. performed a long-term, follow-up retrospective study to address the outcome of patients with E/R(+) leukemia relapses [158]. They reported that E/R had a strong effect on overall survival after relapse. In 81% of cases the relapses that were found late, they were combined with extramedullary relapses. The 5-year survival rate of patients with E/R(+) ALL relapses reached 81% when the relapse occurred after 36 months [158]. In multivariate analysis, only the duration of first remission remained associated with outcome. Overall, they found an excellent outcome for patients with E/R(+) leukemia relapses that occurred more than 36 months after diagnosis [158]. The duration of first complete remission may be a guide to define the treatment strategy for patients with relapsed E/R(+) leukemia. Similar findings were reported by other groups [159-163], demonstrating that E/R fusion is a sign of favorable prognosis although relapse does happen in 20% of patients. The report from Borssen et al. is especially intriguing since t(12; 21)(p13; q22) E/R fusion is associated with hTERT promoter [164] methylation with short telomeres, and thus with more favorable prognosis of ALL.
Current problems and future directions
ETS proteins are frequently activated in human cancers by translocation. To make GEMM for EWS, the EWS-FLI1 fusion transcripts express exclusively in mesenchymal stem cells to avoid its expression in hematological tissues. When crossed with interferon -inducible Mx1-Cre mice, activation of EWS-FLI1 resulted in the rapid development of myeloid/erythroid leukemia characterized by expansion of mononuclear cells causing hepatosplenomegaly, severe anemia, and death. Gene expression profiles of animals were highly similar to those of human Ewing’s sarcoma, suggesting that activation of EWS-FLI1 was the primary event leading to disease in this model. The Cre-inducible EWS-FLI1 mice provide a novel model system to study the contribution of this oncogene to malignant disease in vivo. The disease is accelerated in p16Ink4a-null mice but not Arf-null mice suggesting the critical role of p16INK4a in Ewing’s sarcoma. In ETS family, the ERG and ETV genes are frequently activated by translocation in human cancer; since the fusion protein often has nearly normal or normal functions of ERG, we speculate that overexpression of ERG proteins is essential for prostate cancer development. The prognostic value of TMPRSS2-ERG fusion in human PCa is still controversial; it seems that it is associated with intermediate survival outcomes second to p53 and PTEN inactivation. The prognostic value of TMPRSS2-ERG fusion will be determined in future prospective studies.
In pediatric B-cell ALL, the translocation involving ETV6 and RUNX1 are very common. It is believed that this is a favorable prognostic factor. The fusion protein develops as an early event in childhood ALL. CDKN2a/b, ETV6, PAX5, deletions, -6q, and +16 are among the earliest genetic aberrations in E/R(+) ALL; they are secondary events that contribute to the leukemic disease. Although 20% of patients suffer from late relapse, studies show that relapsed leukemia responds to chemotherapy with 5-year survival rate of more than 80%, and thus it is believed that it is a favorable prognostic indicator. However, leukemia-free survival remains low for many patients after relapse and despite efforts to intensify therapeutic outcomes for these children. The development of novel therapeutic approaches such as anti-sense therapy or small molecule inhibitor to the chimeric gene holds great promise for the first-line treatment for patients with E/R(+) relapsed ALL.
In any case of human cancer with ETS translocations, specific therapy can be attempted to fusion proteins to eradicate tumor cells since they play critical roles in causing the neoplastic disease.
We thank all other members of Dr. Inoue’s lab for sharing unpublished research data.
K. Inoue was supported by NIH/NCI 2R01CA106314, ACS RSG-07-207-01-MGO, and KG080179.
The authors declare no conflicts of interest.
EWS-FLI1 and TMPRSS2-ERG by EAF and KI; ETV6-RUNX1 by AM and KI. English grammar was checked by AM and EAF.
- Bister K, Nunn M, Moscovici C, Perbal B, Baluda M, et al. (1982) Acute leukemia viruses E26 and avian myeloblastosis virus have related transformation-specific RNA sequences but different genetic structures, gene products, and oncogenic properties. Proc Natl Acad Sci U S A 79: 3677-3681. [Crossref]
- Moelling K, Pfaff E, Beug H, Beimling P, Bunte T, et al. (1985) DNA-binding activity is associated with purified myb proteins from AMV and E26 viruses and is temperature-sensitive for E26 ts mutants. Cell 40: 983-990. [Crossref]
- Metz T, Graf T (1991) Fusion of the nuclear oncoproteins v-Myb and v-Ets is required for the leukemogenicity of E26 virus. Cell 66: 95-105. [Crossref]
- Findlay VJ, LaRue AC, Turner DP, Watson PM, Watson DK (2013) Understanding the role of ETS-mediated gene regulation in complex biological processes. Adv Cancer Res 119: 1-61. [Crossref]
- Feldman RJ, Sementchenko VI, Watson DK (2003a) The epithelial-specific Ets factors occupy a unique position in defining epithelial proliferation, differentiation and carcinogenesis. Anticancer Res 23: 2125-2131. [Crossref]
- Feldman RJ, Sementchenko VI, Gayed M, Fraig MM, Watson DK (2003) Pdef expression in human breast cancer is correlated with invasive potential and altered gene expression. Cancer Res 63: 4626-4631. [Crossref]
- Fry EA, Inoue K (2018) ETS1 and ETS2 proteins in human cancer. Cancer Reports and Reviews. In press.
- Sacchi N, Watson DK, Guerts van Kessel AH, Hagemeijer A, Kersey J, et al. (1986) Hu-ets-1 and Hu-ets-2 genes are transposed in acute leukemias with (4;11) and (8;21) translocations. Science 231: 379-382. [Crossref]
- Sacchi N, Cheng SV, Tanzi RE, Gusella JF, Drabkin HA, et al. (1988) The ETS genes on chromosome 21 are distal to the breakpoint of the acute myelogenous leukemia translocation (8;21). Genomics 3: 110-116. [Crossref]
- Nucifora G, Birn DJ, Erickson P, Gao J, LeBeau MM, et al. (1993) Detection of DNA rearrangements in the AML1 and ETO loci and of an AML1/ETO fusion mRNA in patients with t(8;21) acute myeloid leukemia. Blood 81: 883-888. [Crossref]
- Miyoshi H, Kozu T, Shimizu K, Enomoto K, Maseki N, et al. (1993) The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J 12: 2715-2721. [Crossref]
- Rao VN, Papas TS, Reddy ES (1987) erg, a human ets-related gene on chromosome 21: alternative splicing, polyadenylation, and translation. Science 237: 635-639. [Crossref]
- Rao VN, Modi WS, Drabkin HD, Patterson D, O'Brien SJ, et al. (1988) The human erg gene maps to chromosome 21, band q22: relationship to the 8; 21 translocation of acute myelogenous leukemia. Oncogene 3: 497-500. [Crossref]
- Murakami K, Mavrothalassitis G, Bhat NK, Fisher RJ, Papas TS (1993) Human ERG-2 protein is a phosphorylated DNA-binding protein--a distinct member of the ets family. Oncogene 8: 1559-1566. [Crossref]
- Buchanan J, Tirado CA (2016) A t(16;21)(p11;q22) in Acute Myeloid Leukemia (AML) Resulting in Fusion of the FUS/TLS and ERG Genes: A Review of the Literature. J Assoc Genet Technol 42: 24-33. [Crossref]
- Hibshoosh H, Lattes R (1997) Immunohistochemical and molecular genetic approaches to soft tissue tumor diagnosis: a primer. Semin Oncol 24: 515-525. [Crossref]
- Acs B, Szarvas T, Szekely N, Nyirady P, Szasz AM1 (2015) Current State of ERG as Biomarker in Prostatic Adenocarcinoma. Curr Cancer Drug Targets 15: 643-651. [Crossref]
- Berg KD (2016) The prognostic and predictive value of TMPRSS2-ERG gene fusion and ERG protein expression in prostate cancer biopsies. Dan Med J 63. [Crossref]
- Sanguedolce F, Cormio A, Brunelli M, D'Amuri A, Carrieri G, et al. (2016) Urine TMPRSS2: ERG Fusion Transcript as a Biomarker for Prostate Cancer: Literature Review. Clin Genitourin Cancer 14: 117-121. [Crossref]
- Sundaresh A, Williams O (2017) Mechanism of ETV6-RUNX1 Leukemia. Adv Exp Med Biol 962: 201-216. [Crossref]
- Sun C, Chang L, Zhu X (2017) Pathogenesis of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia and mechanisms underlying its relapse. Oncotarget 8: 35445-35459. [Crossref]
- Reddy ES, Rao VN, Papas TS (1987) The erg gene: a human gene related to the ets oncogene. Proc Natl Acad Sci U S A 84: 6131-6135. [Crossref]
- Owczarek CM, Portbury KJ, Hardy MP, O'Leary DA, Kudoh J, et al. (2004) Detailed mapping of the ERG-ETS2 interval of human chromosome 21 and comparison with the region of conserved synteny on mouse chromosome 16. Gene 324: 65-77. [Crossref]
- Ichikawa H, Shimizu K, Hayashi Y, Ohki M (1994) An RNA-binding protein gene, TLS/FUS, is fused to ERG in human myeloid leukemia with t(16;21) chromosomal translocation. Cancer Res 54: 2865-2868. [Crossref]
- Kim SK, Park YK (2016) Ewing sarcoma: a chronicle of molecular pathogenesis. Hum Pathol 55: 91-100. [Crossref]
- Machado I, Navarro S, Llombart-Bosch A (2016) Ewing sarcoma and the new emerging Ewing-like sarcomas: (CIC and BCOR-rearranged-sarcomas). A systematic review. Histol Histopathol 31: 1169-1181. [Crossref]
- Yu H, Ge Y, Guo L, Huang L (2017) Potential approaches to the treatment of Ewing's sarcoma. Oncotarget 8: 5523-5539. [Crossref]
- Downing JR, Head DR, Parham DM, Douglass EC, et al. (1993) Detection of the (11;22)(q24;q12) translocation of Ewing's sarcoma and peripheral neuroectodermal tumor by reverse transcription polymerase chain reaction. Am J Pathol 143: 1294-1300. [Crossref]
- Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, et al. (1994) A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet 6: 146-151. [Crossref]
- Torchia EC, Jaishankar S, Baker SJ (2003) Ewing tumor fusion proteins block the differentiation of pluripotent marrow stromal cells. Cancer Res 63: 3464-3464. [Crossref]
- Torchia EC, Boyd K, Rehg JE, Qu C, Baker SJ (2007) EWS/FLI-1 induces rapid onset of myeloid/erythroid leukemia in mice. Mol Cell Biol 27: 7918-7934. [Crossref]
- Riggi N, Stamenkovic I (2007) The Biology of Ewing sarcoma. Cancer Lett 254: 1-10. [Crossref]
- Cidre-Aranaz F, Alonso J (2015) EWS/FLI1 Target Genes and Therapeutic Opportunities in Ewing Sarcoma. Front Oncol 5: 162. [Crossref]
- Erkizan HV, Scher LJ, Gamble SE, Barber-Rotenberg JS, Sajwan KP, et al. (2011) Novel peptide binds EWS-FLI1 and reduces the oncogenic potential in Ewing tumors. Cell Cycle 10: 3397-3408. [Crossref]
- Inoue K, Roussel MF, Sherr CJ (1999) Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc Natl Acad Sci U S A 96: 3993-3998. [Crossref]
- Schwentner R, Papamarkou T, Kauer MO, Stathopoulos V, Yang F, et al. (2015) EWS-FLI1 employs an E2F switch to drive target gene expression. Nucleic Acids Res 43: 2780-2789. [Crossref]
- Mallakin A, Taneja P, Matise, LA, Willingham MC, Inoue K (2006) Expression of Dmp1 in specific differentiated, nonproliferating cells and its repression by E2Fs. Oncogene 25: 7703-7713. [Crossref]
- Taneja P, Frazier DP, Sugiyama T, Lagedrost SJ, Inoue K (2008) Control of cellular physiology by transcription factors E2F and their roles in ‘Carcinogenesis in Control of Cellular Physiology by E2F transcription factors’. (Edtr: Ken-ichi Yoshida. Research Signpost, Trivandrum, Kerala, India). Chapter 10: 179-197.
- Chen HZ, Tsai SY, Leone G (2009) Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 9: 785-797. [Crossref]
- Thurlings I, de Bruin A (2016) E2F Transcription Factors Control the Roller Coaster Ride of Cell Cycle Gene Expression. Methods Mol Biol 1342: 71-88. [Crossref]
- García-Aragoncillo E, Carrillo J, Lalli E, Agra N, Gómez-López G, et al. (2008) DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell-cycle progression in Ewing's tumor cells. Oncogene 27: 6034-6043. [Crossref]
- Abaan OD, Levenson A, Khan O, Furth PA, Uren A, et al. (2005) TPL1 is a direct transcriptional target of EWS-FLI1 and modulates Ewing's Sarcoma tumorigenesis. Oncogene 24: 2715-2722. [Crossref]
- Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, et al. (2009) GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem 284: 9074-9082. [Crossref]
- Kim S, Denny CT, Wisdom R (2006) Cooperative DNA binding with AP-1 proteins is required for transformation by EWS-Ets fusion proteins. Mol Cell Biol 26: 2467-2478. [Crossref]
- Erkizan HV, Uversky VN, Toretsky JA (2010) Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing's sarcoma. Clin Cancer Res 16: 4077-4083. [Crossref]
- Mosakhani N, Guled M, Leen G, Calabuig-Fariñas S, Niini T, et al. (2012) An integrated analysis of miRNA and gene copy numbers in xenografts of Ewing's sarcoma. J Exp Clin Cancer Res 31: 24. [Crossref]
- Vousden KH, Prives C (2009) Blinded by the Light: The Growing Complexity of p53. Cell 137: 413-431. [Crossref]
- Kruiswijk F, Labuschagne CF, Vousden KH (2015) p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16: 393-405. [Crossref]
- Maggi LB Jr, Winkeler CL, Miceli AP, Apicelli AJ, Brady SN, et al. (2014) ARF tumor suppression in the nucleolus. Biochim Biophys Acta 1842: 831-839. [Crossref]
- Sharpless NE, Sherr CJ (2015) Forging a signature of in vivo senescence. Nat Rev Cancer 15: 397-408. [Crossref]
- Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, et al. (2005) Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J Clin Oncol 23: 548-558. [Crossref]
- Lin PP, Pandey MK, Jin F, Xiong S, Deavers M, et al. (2008) EWS-FLI1 induces developmental abnormalities and accelerates sarcoma formation in a transgenic mouse model. Cancer Res 68: 8968-8975. [Crossref]
- Deneen B, Denny CT (2001) Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene 20: 6731-6741. [Crossref]
- Inoue K, Sherr CJ (1998) Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol 18: 1590-1600. [Crossref]
- Inoue K, Sherr CJ, Shapiro LH (1998) Regulation of the CD13/aminopeptidase N gene by DMP1, a transcription factor antagonized by D-type cyclins. J Biol Chem 273: 29188-29194. [Crossref]
- Inoue K, Wen R, Rehg JE, Adachi M, Cleveland JL, et al. (2000) Disruption of the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev 14: 1797-1809. [Crossref]
- Inoue K, Zindy F, Randle DH, Rehg JE, Sherr CJ (2001) Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev 15: 2934-2939. [Crossref]
- Sreeramaneni R, Chaudhry A, McMahon M, Sherr CJ, Inoue K (2005) Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol 25: 220-232. [Crossref]
- Taneja P, Mallakin A, Matise LA, Frazier DP, Choudhary M, et al. (2007) Repression of Dmp1 and Arf transcription by anthracyclins: critical roles of the NF-kappaB subunit p65. Oncogene 26: 7457-7466. [Crossref]
- Mallakin A, Sugiyama T, Taneja P, Matise LA, Frazier DP, et al. (2007) Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell 12: 381-394. [Crossref]
- Mallakin A, Sugiyama T, Kai F, Taneja P, Kendig RD, et al. (2010) The Arf-inducing transcription factor Dmp1 encodes transcriptional activator of amphiregulin, thrombospondin-1, JunB and Egr1. Int J Cancer 126: 1403-1416. [Crossref]
- Frazier DP, Kendig RD, Kai F, Maglic D, Sugiyama T, et al. (2012) Dmp1 physically interacts with p53 and positively regulates p53’s stabilization, nuclear localization, and function. Cancer Res 72: 1740-1750. [Crossref]
- Maglic D, Zhu S, Fry EA, Taneja P, Kai F, et al. (2013) Prognostic value of the hDMP1-ARF-Hdm2-p53 pathway in breast cancer. Oncogene 32: 4120-4129. [Crossref]
- Zhu S, Mott RT, Fry EA, Taneja P, Kulik G, et al. (2013) Cooperation between Dmp1 loss and cyclin D1 overexpression in breast cancer. Am J Pathol 183: 1339-1350. [Crossref]
- Fry EA, Taneja P, Maglic D, Zhu S, Sui G, et al. (2013) Dmp1α inhibits HER2/neu-induced mammary tumorigenesis. PLoS One 8: e77870. [Crossref]
- Maglic D, Stovall DB, Cline JM, Fry EA, Mallakin A, et al. (2015) DMP1β, a splice isoform of the tumour suppressor DMP1 locus, induces proliferation and progression of breast cancer. J Pathol 236: 90-102. [Crossref]
- Tschan MP, Federzoni EA, Haimovici A, Britschgi C, Moser BA, et al. (2015) Human DMTF1ß antagonizes DMTF1a regulation of the p14(ARF) tumor suppressor and promotes cellular proliferation. Biochim Biophys Acta 1849: 1198-1208. [Crossref]
- Kendig RD, Kai F, Fry EA, Inoue K (2017) Stabilization of the p53-DNA Complex by the Nuclear Protein Dmp1α. Cancer Invest 35: 301-312. [Crossref]
- Inoue K, Mallakin A, Frazier DP (2007) Dmp1 and tumor suppression. Oncogene 26: 4329-4335. [Crossref]
- Inoue K, Fry EA, Frazier DP (2016) Transcription factors that interact with p53 and Mdm2. Int J Cancer 138: 1577-1585. [Crossref]
- Inoue K, Fry EA (2016) Aberrant splicing of the DMP1-ARF-MDM2-p53 pathway in cancer. Int J Cancer 139: 33-41. [Crossref]
- Fry EA, Taneja P, Inoue K (2017) Oncogenic and tumor-suppressive mouse models for breast cancer engaging HER2/neu. Int J Cancer 140: 495-503. [Crossref]
- Inoue K, Fry EA (2017) Haploinsufficient tumor suppressor genes. Adv Med Biol 118: 83-122. [Crossref]
- Aryee DN, Niedan S, Kauer M, Schwentner R, Bennani-Baiti IM, et al. (2010) Hypoxia modulates EWS-FLI1 transcriptional signature and enhances the malignant properties of Ewing's sarcoma cells in vitro. Cancer Res 70: 4015-4023. [Crossref]
- Taneja P, Zhu S, Maglic D, Fry EA, Kendig RD, et al. (2011) Transgenic and knockout mice models to reveal the functions of tumor suppressor genes. Clin Med Insights Oncol 5: 235-257. [Crossref]
- Inoue K, Fry EA, Taneja P (2013) Recent progress in mouse models for tumor suppressor genes and its implications in human cancer. Clin Med Insights Oncol 7: 103-122. [Crossref]
- Riggi N, Suvà ML, Suvà D, Cironi L, Provero P, et al. (2008) EWS-FLI-1 expression triggers a Ewing's sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res 68: 2176-2185. [Crossref]
- Torchia EC, Boyd K, Rehg JE, Qu C, Baker SJ (2007) EWS/FLI-1 induces rapid onset of myeloid/erythroid leukemia in mice. Mol Cell Biol 27: 7918-7934. [Crossref]
- Velasco-Hernandez T, Säwén P, Bryder D, Cammenga J (2016) Potential Pitfalls of the Mx1-Cre System: Implications for Experimental Modeling of Normal and Malignant Hematopoiesis. Stem Cell Reports 7: 11-18. [Crossref]
- Elzi DJ, Song M, Houghton PJ, Chen Y, Shiio Y (2015) The role of FLI-1-EWS, a fusion gene reciprocal to EWS-FLI-1, in Ewing sarcoma. Genes Cancer 6: 452-461. [Crossref]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, et al. (2005) Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310: 644-648. [Crossref]
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, et al. (2007) Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448: 595-599. [Crossref]
- Williams JL, Yoshimoto M, Boag AH, Squire JA, Park, PC (2011) TMPRSS2:ETS gene fusions in prostate cancer. Atlas Genet Cytogenet Oncol Haematol 15: 699-716.
- Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, et al. (2010) An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17: 443-454. [Crossref]
- Cancer Genome Atlas Research Network (2015) The Molecular Taxonomy of Primary Prostate Cancer. Cell 163: 1011-1025. [Crossref]
- Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, et al. (2009) Induced chromosomal proximity and gene fusions in prostate cancer. Science 326: 1230. [Crossref]
- Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, et al. (2008) Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 10: 177-188. [Crossref]
- Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, et al. (2009) Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet 41: 619-624. [Crossref]
- King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, et al. (2009) Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet 41: 524-526. [Crossref]
- Mounir Z, Lin F, Lin VG, Korn JM, Yu Y, et al. (2015) TMPRSS2:ERG blocks neuroendocrine and luminal cell differentiation to maintain prostate cancer proliferation. Oncogene 34: 3815-3825. [Crossref]
- Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, et al. (2006) TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res 66: 8337-8341. [Crossref]
- Lapointe J, Kim YH, Miller MA, Li C, Kaygusuz G, et al. (2007) A variant TMPRSS2 isoform and ERG fusion product in prostate cancer with implications for molecular diagnosis. Mod Pathol 20: 467-473. [Crossref]
- Wang J, Cai Y, Ren C, Ittmann M (2006) Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res 66: 8347-8351. [Crossref]
- Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, et al. (2009) ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A 106: 12465-12470. [Crossref]
- Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J, et al. (2010) Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc Natl Acad Sci U S A 107: 2610-2615. [Crossref]
- Fallahabadi ZR, Noori Daloii MR, Mahdian R, Behjati F, Shokrgozar MA, et al. (2016) Frequency of PTEN alterations, TMPRSS2-ERG fusion and their association in prostate cancer. Gene 575: 755-760. [Crossref]
- Bismar TA, Yoshimoto M, Vollmer RT, Duan Q, Firszt M, et al. (2011) PTEN genomic deletion is an early event associated with ERG gene rearrangements in prostate cancer. BJU Int 107: 477-485. [Crossref]
- Krohn A, Diedler T, Burkhardt L, Mayer PS, De Silva C, et al. (2012) Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am J Pathol 181: 401-412. [Crossref]
- Burdelski C, Ruge OM, Melling N, Koop C, Simon R, et al. (2015) HDAC1 overexpression independently predicts biochemical recurrence and is associated with rapid tumor cell proliferation and genomic instability in prostate cancer. Exp Mol Pathol 98: 419-426. [Crossref]
- Schweizer L, Rizzo CA, Spires TE, Platero JS, Wu Q, et al. (2008) The androgen receptor can signal through Wnt/beta-Catenin in prostate cancer cells as an adaptation mechanism to castration levels of androgens. BMC Cell Biol 9: 4. [Crossref]
- Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, et al. (2008) TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene 27: 5348-5353. [Crossref]
- Hawksworth D, Ravindranath L, Chen Y, Furusato B, Sesterhenn IA, et al. (2010) Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. Prostate Cancer Prostatic Dis 13: 311-315. [Crossref]
- Zhang H, Shang YP, Chen HY, Li J (2017) Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol Res 47: 149-159. [Crossref]
- Zhang S, Li Y, Wu Y, Shi K, Bing L, et al. (2012) Wnt/β-catenin signaling pathway upregulates c-Myc expression to promote cell proliferation of P19 teratocarcinoma cells. Anat Rec (Hoboken) 295: 2104-2113. [Crossref]
- Demichelis F, Fall K, Perner S, Andren O, Schmidt F, et al. (2007) TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 26: 4596-4599. [Crossref]
- Attard G, Clark J, Ambroisine L, Fisher G, Kovacs G, et al. (2008) Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 27: 253-263. [Crossref]
- Markert EK, Mizuno H, Vazquez A, Levine AJ (2011) Molecular classification of prostate cancer using curated expression signatures. Proc Natl Acad Sci U S A 108: 21276-21281. [Crossref]
- Font-Tello A, Juanpere N, de Muga S, Lorenzo M, Lorente JA, et al. (2015) Association of ERG and TMPRSS2-ERG with grade, stage, and prognosis of prostate cancer is dependent on their expression levels. Prostate 75: 1216-1226. [Crossref]
- Hernández S, Font-Tello A, Juanpere N, de Muga S, Lorenzo M, et al. (2016) Concurrent TMPRSS2-ERG and SLC45A3-ERG rearrangements plus PTEN loss are not found in low grade prostate cancer and define an aggressive tumor subset. Prostate 76: 854-865. [Crossref]
- Deplus R, Delliaux C, Marchand N, Flourens A, Vanpouille N, et al. (2017) TMPRSS2-ERG fusion promotes prostate cancer metastases in bone. Oncotarget 8: 11827-11840. [Crossref]
- Saramäki OR, Harjula AE, Martikainen PM, Vessella RL, Tammela TL, et al. (2008) TMPRSS2:ERG fusion identifies a subgroup of prostate cancers with a favorable prognosis. Clin Cancer Res 14: 3395-3400. [Crossref]
- Hermans KG, Boormans JL, Gasi D, van Leenders GJ, Jenster G, et al. (2009) Overexpression of prostate-specific TMPRSS2(exon 0)-ERG fusion transcripts corresponds with favorable prognosis of prostate cancer. Clin Cancer Res 15: 6398-6403. [Crossref]
- Fleischmann A, Saramäki OR, Zlobec I, Rotzer D, Genitsch V, et al. (2014) Prevalence and prognostic significance of TMPRSS2-ERG gene fusion in lymph node positive prostate cancers. Prostate 74: 1647-1654. [Crossref]
- FitzGerald LM, Agalliu I, Johnson K, Miller MA, Kwon EM, et al. (2008) Association of TMPRSS2-ERG gene fusion with clinical characteristics and outcomes: results from a population based study of prostate cancer. BMC Cancer 8: 230. [Crossref]
- Gopalan A, Leversha MA, Satagopan JM, Zhou Q, Al-Ahmadie HA, et al. (2009) TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res 69: 1400-1406. [Crossref]
- Minner S, Enodien M, Sirma H, Luebke AM, Krohn A, et al. (2011) ERG status is unrelated to PSA recurrence in radically operated prostate cancer in the absence of antihormonal therapy. Clin Cancer Res 17: 5878-5888. [Crossref]
- Toubaji A, Albadine R, Meeker AK, Isaacs WB, Lotan T, et al. (2011) Increased gene copy number of ERG on chromosome 21 but not TMPRSS2-ERG fusion predicts outcome in prostatic adenocarcinomas. Mod Pathol 24: 1511-1520. [Crossref]
- Adamo P, Ladomery MR (2016) The oncogene ERG: a key factor in prostate cancer. Oncogene 35: 403-414. [Crossref]
- Tapia-Laliena MA, Korzeniewski N, Hohenfellner M, Duensing S (2014) High-risk prostate cancer: a disease of genomic instability. Urol Oncol 32: 1101-1107. [Crossref]
- Danielsen HE, Pradhan M, Novelli M (2016) Revisiting tumour aneuploidy - the place of ploidy assessment in the molecular era. Nat Rev Clin Oncol 13: 291-304. [Crossref]
- Frost BM, Forestier E, Gustafsson G, Nygren P, Hellebostad M, et al. (2004) Translocation t(12;21) is related to in vitro cellular drug sensitivity to doxorubicin and etoposide in childhood acute lymphoblastic leukemia. Blood 104: 2452-2457. [Crossref]
- Zuna J, Madzo J, Krejci O, Zemanova Z, Kalinova M, et al. (2011) ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood 117: 368-369. [Crossref]
- Sun C, Chang L, Zhu X (2017) Pathogenesis of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia and mechanisms underlying its relapse. Oncotarget 8: 35445-35459. [Crossref]
- Pais AP, Amare Kadam PS, Raje GC, Banavali S, Parikh P, et al. (2008) RUNX1 aberrations in ETV6/RUNX1-positive and ETV6/RUNX1-negative patients: its hemato-pathological and prognostic significance in a large cohort (619 cases) of ALL. Pediatr Hematol Oncol 25: 582-597. [Crossref]
- Bokemeyer A, Eckert C, Meyr F, Koerner G, von Stackelberg A, et al. (2014) Copy number genome alterations are associated with treatment response and outcome in relapsed childhood ETV6/RUNX1-positive acute lymphoblastic leukemia. Haematologica 99: 706-714. [Crossref]
- Janczar K, Janczar S, Pastorczak A, Mycko K, Paige AJ, et al. (2015) Preserved global histone H4 acetylation linked to ETV6-RUNX1 fusion and PAX5 deletions is associated with favorable outcome in pediatric B-cell progenitor acute lymphoblastic leukemia. Leuk Res 39: 1455-1461. [Crossref]
- Gandemer V, Rio AG, de Tayrac M, Sibut V, Mottier S, et al. (2007) Five distinct biological processes and 14 differentially expressed genes characterize TEL/AML1-positive leukemia. BMC Genomics 8: 385. [Crossref]
- Lee JW, Kim SK, Jang PS, Chung NG, Jeong DC, et al. (2017) Outcome and Prognostic Factors for ETV6/RUNX1 Positive Pediatric Acute Lymphoblastic Leukemia Treated at a Single Institution in Korea. Cancer Res Treat 49: 446-453. [Crossref]
- Sun C, Chang L, Zhu X (2017) Pathogenesis of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia and mechanisms underlying its relapse. Oncotarget 8: 35445-35459. [Crossref]
- Maia AT, Koechling J, Corbett R, Metzler M, Wiemels JL, et al. (2004) Protracted postnatal natural histories in childhood leukemia. Genes Chromosomes Cancer 39: 335-340. [Crossref]
- Fischer M, Schwieger M, Horn S, Niebuhr B, Ford A, et al. (2005) Defining the oncogenic function of the TEL/AML1 (ETV6/RUNX1) fusion protein in a mouse model. Oncogene 24: 7579-7591. [Crossref]
- Raynaud S, Cave H, Baens M, Bastard C, Cacheux V, et al. (1996) The 12;21 translocation involving TEL and deletion of the other TEL allele: two frequently associated alterations found in childhood acute lymphoblastic leukemia. Blood 87: 2891-2899. [Crossref]
- Lopez RG, Carron C, Oury C, Gardellin P, Bernard O, et al. (1999) TEL is a sequence-specific transcriptional repressor. J Biol Chem 274: 30132-30138. [Crossref]
- Coniat MB, Poirel H, Leblanc T, Bernard OA, Berger R (1999) Loss of the TEL/ETV6 gene by a second translocation in ALL patients with t(12;21). Leuk Res 23: 895-899. [Crossref]
- Mrózek K, Harper DP, Aplan PD (2009) Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol Oncol Clin North Am 23: 991-1010, v. [Crossref]
- Al-Shehhi H, Konn ZJ, Schwab CJ, Erhorn A, Barber KE, et al. (2013) Abnormalities of the der(12)t(12;21) in ETV6-RUNX1 acute lymphoblastic leukemia. Genes Chromosomes Cancer 52: 202-213. [Crossref]
- Borst L, Wesolowska A, Joshi T, Borup R, Nielsen FC, et al. (2012) Genome-wide analysis of cytogenetic aberrations in ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia. Br J Haematol 157: 476-482. [Crossref]
- Wall M, Rayeroux KC, MacKinnon RN, Zordan A, Campbell LJ (2012) ETV6 deletion is a common additional abnormality in patients with myelodysplastic syndromes or acute myeloid leukemia and monosomy 7. Haematologica 97: 1933-1936. [Crossref]
- Inoue K, Fry EA (2018) Aberrant expression of p16INK4a in human cancers - a new biomarker? Cancer Rep Rev 2. [Crossref]
- Inoue K, Fry EA (2018) Overexpression of ARF in human cancer: a new biomarker? Tumor Microenviron. [In press]. [Crossref]
- Sulong S, Moorman AV, Irving JA, Strefford JC, Konn ZJ, et al. (2009) A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood 113: 100-107. [Crossref]
- Janczar K, Janczar S, Pastorczak A, Mycko K, Paige AJ, et al. (2015) Preserved global histone H4 acetylation linked to ETV6-RUNX1 fusion and PAX5 deletions is associated with favorable outcome in pediatric B-cell progenitor acute lymphoblastic leukemia. Leuk Res 39: 1455-1461. [Crossref]
- Lilljebjörn H, Soneson C, Andersson A, Heldrup J, Behrendtz M, et al. (2010) The correlation pattern of acquired copy number changes in 164 ETV6/RUNX1-positive childhood acute lymphoblastic leukemias. Hum Mol Genet 19: 3150-3158. [Crossref]
- Fuka G, Kauer M, Kofler R, Haas OA, Panzer-Grümayer R (2011) The leukemia-specific fusion gene ETV6/RUNX1 perturbs distinct key biological functions primarily by gene repression. PLoS One 6: e26348. [Crossref]
- Cooper SL, Brown PA (2015) Treatment of pediatric acute lymphoblastic leukemia. Pediatr Clin North Am 62: 61-73. [Crossref]
- Locatelli F, Schrappe M, Bernardo ME, Rutella S (2012) How I treat relapsed childhood acute lymphoblastic leukemia. Blood 120: 2807-2816. [Crossref]
- Bertaina A, Vinti L, Strocchio L, Gaspari S, Caruso R, et al. (2017) The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br J Haematol 176: 629-636. [Crossref]
- Welborn JL (1994) Impact of reinduction regimens for relapsed and refractory acute lymphoblastic leukemia in adults. Am J Hematol 45: 341-344. [Crossref]
- Raetz EA, Bhatla T (2012) Where do we stand in the treatment of relapsed acute lymphoblastic leukemia? Hematology Am Soc Hematol Educ Program 2012: 129-136. [Crossref]
- Bhojwani D, Pui CH (2013) Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol 14: 205-217. [Crossref]
- Fry TJ, Mackall CL (2013) T-cell adoptive immunotherapy for acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program 2013: 348-353. [Crossref]
- Konrad M, Metzler M, Panzer S, Ostreicher I, Peham M, et al. (2003) Late relapses evolve from slow-responding subclones in t(12;21)-positive acute lymphoblastic leukemia: evidence for the persistence of a preleukemic clone. Blood 101: 3635-3640. [Crossref]
- Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, et al. (2008) Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322: 1377-1380. [Crossref]
- Panzer-Grumayer ER, Cazzaniga G, van der Velden VH, del Giudice L, Peham M, et al. (2005) Immunogenotype changes prevail in relapses of young children with TEL-AML1-positive acute lymphoblastic leukemia and derive mainly from clonal selection. Clin Cancer Res 11: 7720-7727. [Crossref]
- Zuna J, Ford AM, Peham M, Patel N, Saha V, et al. (2004) TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res 10: 5355-5360. [Crossref]
- Lawson SE, Harrison G, Richards S, Oakhill A, Stevens R, et al. (2000) The UK experience in treating relapsed childhood acute lymphoblastic leukaemia: a report on the medical research council UKALLR1 study. Br J Haematol 108: 531-543. [Crossref]
- Kuster L, Grausenburger R, Fuka G, Kaindl U, Krapf G, et al. (2011) ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood 117: 2658-2667. [Crossref]
- Gandemer V, Chevret S, Petit A, Vermylen C, Leblanc T, et al. (2012) Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97: 1743-1750. [Crossref]
- Ribera J, Zamora L, Morgades M, Mallo M, Solanes N, et al. (2017) Copy number profiling of adult relapsed B-cell precursor acute lymphoblastic leukemia reveals potential leukemia progression mechanisms. Genes Chromosomes Cancer 56: 810-820. [Crossref]
- van Delft FW, Horsley S, Colman S, Anderson K, Bateman C, et al. (2011) Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117: 6247-6254. [Crossref]
- Moorman AV, Enshaei A, Schwab C, Wade R, Chilton L, et al. (2014) A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood 124: 1434-1444. [Crossref]
- Harrison CJ (2013) Targeting signaling pathways in acute lymphoblastic leukemia: new insights. Hematology Am Soc Hematol Educ Program 2013: 118-125. [Crossref]
- Moorman AV (2016) New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica 101: 407-416. [Crossref]
- Borssén M, Cullman I, Norén-Nyström U, Sundström C, Porwit A, et al. (2011) hTERT promoter methylation and telomere length in childhood acute lymphoblastic leukemia: associations with immunophenotype and cytogenetic subgroup. Exp Hematol 39: 1144-1151. [Crossref]
- Oettgen P (2006) Regulation of vascular inflammation and remodeling by ETS factors. Circ Res 99: 1159-1166. [Crossref]
- Gutierrez-Hartmann A, Duval DL, Bradford AP (2007) ETS transcription factors in endocrine systems. Trends Endocrinol Metab 18: 150-158. [Crossref]
- Stinson J, Inoue T, Yates P, Clancy A, Norton JD, et al. (2003) Regulation of TCF ETS-domain transcription factors by helix-loop-helix motifs. Nucleic Acids Res 31: 4717-4728. [Crossref]
- Huret JL, Bernheim A (1997) t(12;21)(p12;q22). Atlas Genet Cytogenet Oncol Haematol 1: 19-20
- Wiemels JL, Greaves M (1999) Structure and possible mechanisms of TEL-AML1 gene fusions in childhood acute lymphoblastic leukemia. Cancer Res 59: 4075-4082. [Crossref]